


Содержание
Перейти к:
 Наталия Андреевна Сдвигова,
Лейла Ахатовна Гандаева,
Оксана Валерьевна Глоба,
Ирина Вячеславовна Сильнова,
Юлия Игоревна Давыдова,
Елена Юрьевна Басаргина,
Кирилл Викторович Савостьянов
Наталия Андреевна Сдвигова,
Лейла Ахатовна Гандаева,
Оксана Валерьевна Глоба,
Ирина Вячеславовна Сильнова,
Юлия Игоревна Давыдова,
Елена Юрьевна Басаргина,
Кирилл Викторович Савостьянов https://doi.org/10.46563/2686-8997-2025-6-4-221-227
EDN: gdebmq
Перейти к:
Введение. Дефицит окислительного фосфорилирования, 10 тип является редким вариантом митохондриального заболевания, характеризующимся лактат-ацидозом, поражением сердечно-сосудистой и центральной нервной систем. Учитывая вариабельность фенотип-генотипических корреляций, подробное описание клинической картины заболевания является значимым для понимания возможных вариантов течения заболевания.
Материалы и методы. При проведении молекулярно-генетического исследования 314 пробандам детского возраста с направляющим диагнозом «Гипертрофическая кардиомиопатия» выявлен один пациент с дефицитом окислительного фосфорилирования, 10 тип.
Результаты. Проведён анализ характера течения заболевания за период динамического наблюдения (в течение 3 лет).
Заключение. Дефицит окислительного фосфорилирования, 10 тип является редкой причиной гипертрофического фенотипа кардиомиопатии, в литературе описаны единичные случаи данного заболевания, что делает наблюдение значимым для широкого круга специалистов (педиатров, неврологов, кардиологов).
Соблюдение этических стандартов. Получено добровольное информированное согласие от законного представителя пациента на публикацию материала, относящегося к пациенту, в российских и зарубежных периодических медицинских изданиях (от 01.10.2024).
Участие авторов:
Сдвигова Н.А — концепция и дизайн исследования, сбор и обработка материала, написание текста, редактирование;
Гандаева Л.А. — редактирование;
Глоба О.В. — редактирование;
Сильнова И.В. — редактирование;
Давыдова Ю.И. — редактирование;
Басаргина Е.Ю. — редактирование;
Савостьянов К.В. — концепция и дизайн исследования.
Все авторы — утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Поступила 18.08.2025
Принята к печати 30.09.2025
Опубликована 31.01.2026
Сдвигова Н.А., Гандаева Л.А., Глоба О.В., Сильнова И.В., Давыдова Ю.И., Басаргина Е.Ю., Савостьянов К.В. Дефицит окислительного фосфорилирования, 10 тип — редкая причина гипертрофического фенотипа кардиомиопатии. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):221-227. https://doi.org/10.46563/2686-8997-2025-6-4-221-227. EDN: gdebmq
Sdvigova N.A., Gandaeva L.A., Globa O.V., Silnova I.V., Davydova Yu.I., Basargina E.Yu., Savostyanov K.V. Oxidative phosphorylation deficiency type 10 is a rare cause of hypertrophic cardiomyopathy. L.O. Badalyan Neurological Journal. 2025;6(4):221-227. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-221-227. EDN: gdebmq
Введение
Митохондриальные заболевания — это гетерогенная группа наследственных болезней, обусловленная нарушением процессов окислительного фосфорилирования [1, 2].
Дефицит окислительного фосфорилирования, тип 10 (ДОФ-10) был впервые описан в 2012 г. у 2 сибсов с лактат-ацидозом, тяжёлой гипогликемией и гипертрофической кардиомиопатией, рождённых от некровнородственного брака в итальянской семье. Данное заболевание обусловлено наличием патогенных нуклеотидных вариантов в гене MTO1 (mitochondrial tRNA translation optimization), расположенном на длинном плече хромосомы 6 в участке 6q13. Ген MTO1 кодирует белок оптимизации трансляции митохондриальной тРНК 1, являющийся высококонсервативным и экспрессирующимся в тканях с высоким энергопотреблением. Данный белок представляет одну из двух субъединиц фермента, катализирующего 5-карбоксиметиламинометилирование уридинового основания митохондриальной тРНК, специфичной для лейцина, триптофана, глутамина, глутаминовой кислоты и лизина. Эта посттранскрипционная модицикация критически важна для точности и эффективности трансляции митохондриальной ДНК [3–6].
Типичными клиническими проявлениями ДОФ-10 являются лактат-ацидоз, гипертрофическая кардиомиопатия, задержка психомоторного развития, атаксия, судорожные приступы, атрофия зрительных нервов. При проведении магнитно-резонансной томографии (МРТ) головного мозга у некоторых пациентов выявлялись изменения на уровне базальных ганглиев и ножек мозжечка, гипоплазия мозолистого тела, повышение уровня пика лактата по данным МР-спектроскопии. Следует отметить, что генотип-фенотипические корреляции изучены недостаточно [7], а клиническая картина в каждом случае вариабельна.
На момент написания публикации описания ДОФ-10 у детей в отечественной литературе нет, что делает данное наблюдение уникальным и значимым.
Материалы и методы
На базе лаборатории медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России (далее — Центр) с 2019 по 2024 г. проведена молекулярно-генетическая диагностика 314 пробандам детского возраста с направляющим диагнозом «Гипертрофическая кардиомиопатия», регулярно наблюдающимся на базе кардиологического отделения Центра. В качестве причины заболевания у 170 (54%) детей верифицированы нуклеотидные варианты в саркомерных и несаркомерных генах, у 73 (23%) — в генах RAS-патий, у 36 (11,5%) — в генах, ассоциированных с развитием наследственных болезней обмена, у 36 (11,5%) достоверную причину ремоделирования выявить не удалось.
Исследование включало в себя сбор анамнеза с оценкой наследственности, клинический осмотр, лабораторную диагностику (общий анализ крови, биохимический анализ крови, исследование уровня лактата и аммиака), инструментальные методы исследования (эхокардиография (ЭхоКГ), электрокардиография (ЭКГ), суточное мониторирование ЭКГ, МРТ). Всем пациентам проведено молекулярно-генетическое исследование с помощью секвенирования нового поколения — метода выбора для диагностики наследственных заболеваний [8]. В случаях подозрения на вовлечение центральной нервной системы осуществлялась запись на системе видео-ЭЭГ-мониторинга «Nicolet» с использованием международной системы наложения электродов «10-20», с применением дополнительных электродов ЭКГ, МРТ головного мозга на аппарате «GE 3T Discovery 750».
Результаты
В ходе проведения исследования у 6 (19%) из 36 детей с наследственными болезнями обмена верифицированы нуклеотидные варианты в генах, приводящие к митохондриальным нарушениям: ACAD9 (n = 3), ELAC2 (n = 1), POLG (n = 1) и MTO1 (n = 1) [9–11]. В данной статье представлены особенности течения заболевания, сопровождающегося гипертрофией миокарда, при ДОФ-10.
Мальчик Т. от 2-й беременности (1-я — медицинский аборт), протекавшей на фоне токсикоза, уреаплазмоза, кольпита, от 1-х самостоятельных родов в срок, вес при рождении 3740 г, длина 54 см, оценка по шкале Aпгар 8/9 баллов. Период новорождённости протекал без особенностей. Ребёнок наблюдался неврологом с рождения по поводу перинатального поражения центральной нервной системы, синдрома двигательных нарушений, затем — в связи с общим недоразвитием речи; проходил регулярные обследования у офтальмолога по поводу миопии слабой степени, сложного гиперметропического астигматизма, проводилась очковая коррекция.
В 4 мес выявлен умеренный стеноз лёгочной артерии с градиентом 24 мм рт. ст., по поводу которого регулярно наблюдался кардиологом по месту жительства. С 10 лет 8 мес у ребёнка появились жалобы на плохую переносимость физических нагрузок (бег или езда на велосипеде), что проявлялось резким ухудшением самочувствия, рвотой, слабостью, а улучшение наступало после приёма сладкого чая и сна, в этот же период при проведении планового ЭхоКГ отмечено появление умеренной симметричной гипертрофии миокарда, в связи с чем ребёнок был госпитализирован в кардиологическое отделение ФГАУ «НМИЦ здоровья детей» Минздрава России.
При клиническом осмотре в Центре в 11 лет обращало на себя внимание низкое, дисгармоничное физическое развитие за счёт дефицита массы тела (рост 133 см, вес 23 кг, индекс массы тела 13). Анализ лабораторных данных показал повышение уровня лактата в сыворотке крови до 3,7 ммоль/л (референсные значения — 0,5–1,6 ммоль/л), при этом исследование спектра ацилкарнитинов и аминокислот в сухих пятнах крови методом тандемной масс-спектрометрии патологии не выявило. По данным ЭхоКГ подтверждена гипертрофия межжелудочковой перегородки 17 мм и задней стенки левого желудочка 14 мм с минимальными признаками внутрижелудочковой обструкции (градиент 12 мм рт. ст.) и нарушением диастолической функции по 2-му типу (рис. 1).
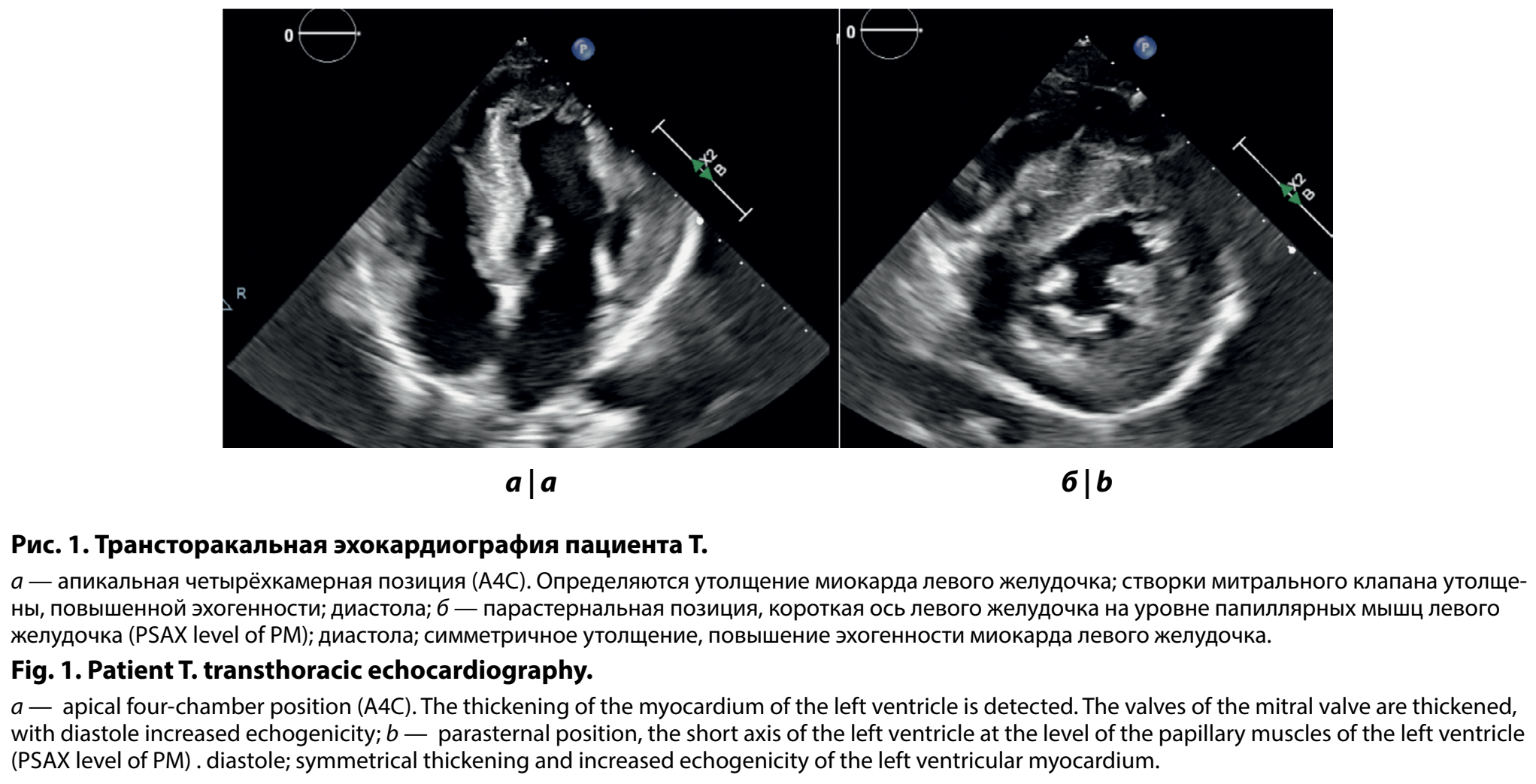
При проведении суточного мониторирования ЭКГ зарегистрирована тахикардия в течение суток (средняя ЧСС днем 118 уд/мин, максимальная 187 уд/мин) без эктопической активности и пауз ритма. Для оценки структуры миокарда была проведена МРТ сердца с контрастированием гадолинием, по результатам которой фиброзных изменений в миокарде не выявлено, максимальная гипертрофия определена в среднем отделе переднеперегородочного и нижнеперегородочного сегментов с признаками начальной обструкции выводных отделов левого желудочка. В ходе проведения теста 6-минутной ходьбы обращено внимание на жалобы пациента на боли в ногах и усталость, при этом ЧСС наросла до 123 уд/мин, артериальное давление — до 140/100 мм рт. ст., субъективная оценка одышки составила 4 балла по шкале Борга.
Принимая во внимание нетипичное течение заболевания, а именно сочетание гипертрофии миокарда без обструкции выходного тракта, снижение толерантности к физическим нагрузкам, боли в нижних конечностях и утомляемость при ходьбе, был заподозрен вторичный генез изменений со стороны сердца, в том числе наследственные болезни обмена. Проведено молекулярно-генетическое обследование в объёме секвенирования клинического экзома с последующей валидацией выявленных вариантов методом прямого автоматического секвенирования по Сэнгеру и проведением сегрегационного анализа в семье. По результатам обследований выявлены нуклеотидные варианты в компаунд-гетерозиготном состоянии в гене MTO1 c.956A>T, p.Glu319Val (унаследован от матери) и c.1430G>A, p.Arg477His (унаследован от отца). Таким образом, был диагностирован комбинированный ДОФ-10 (OMIM 614702).
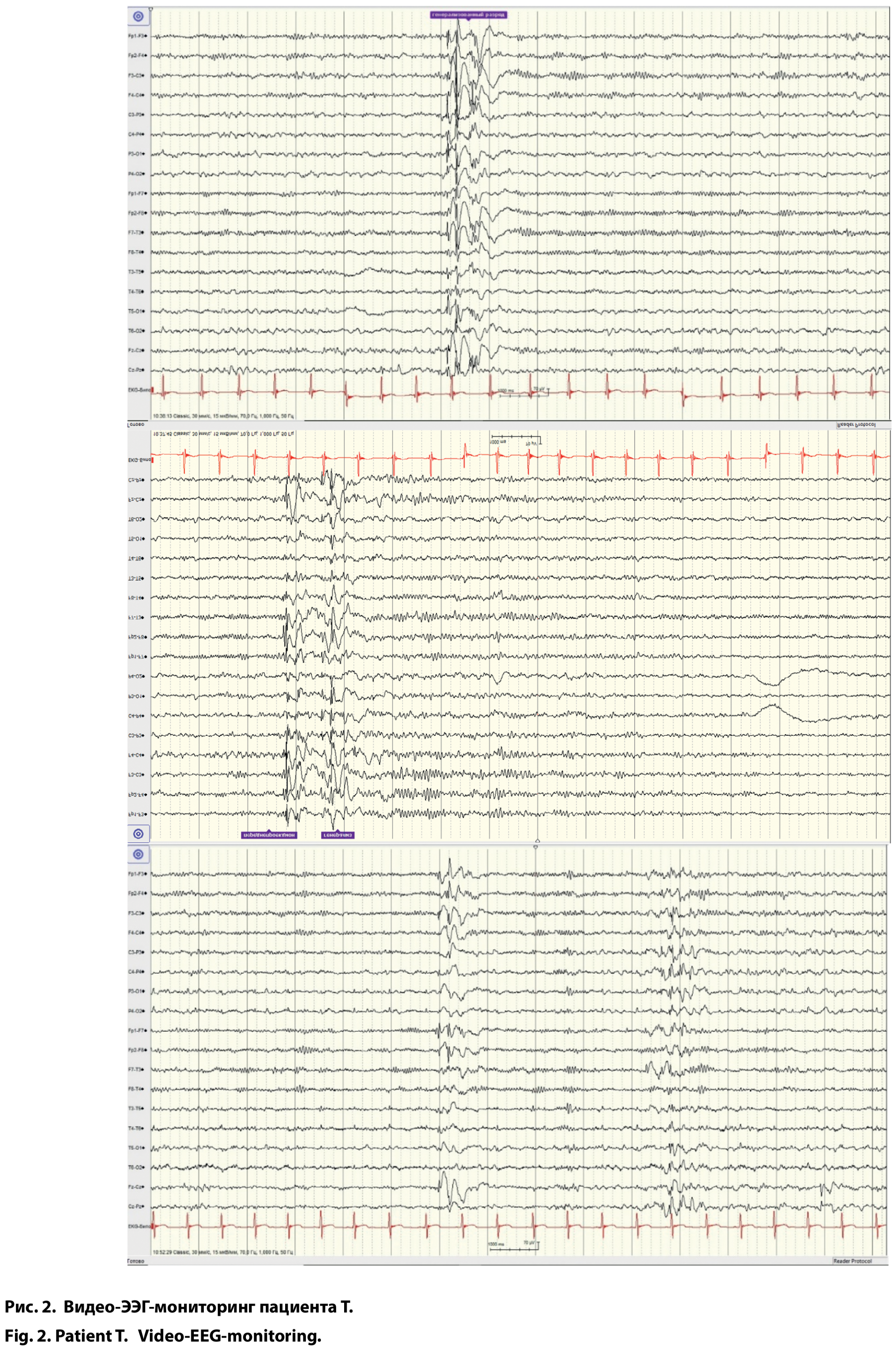
Учитывая потенциальный риск развития судорожных приступов в рамках течения основного заболевания, в 14 лет 2 мес мальчик был обследован в отделении психоневрологии и нейрореабилитации Центра детской психоневрологии ФГАУ «НМИЦ здоровья детей» Минздрава России. В неврологическом статусе отмечены умеренные нарушения когнитивных функций в виде замедления темпа речи и мышления, диффузная мышечная гипотония со снижением мышечной силы, преимущественно в проксимальных отделах конечностей до 3 баллов, интенция при выполнении пальце-носовой пробы, умеренная статическая туловищная атаксия. При проведении видео-ЭЭГ-мониторинга (в течение часа в состоянии бодрствования с выполнением функциональных проб и во время физиологического сна) в периоды бодрствования и поверхностной стадии сна отмечено усиление быстроволновой активности, зарегистрирована мультирегиональная эпилептиформная активность с индексом представленности в период бодрствования 5–10%, во сне — 20%, преимущественно за счёт генерализованных разрядов (рис. 2). Учитывая отсутствие приступов, ребёнок в настоящее время не нуждается в назначении противосудорожной терапии, требуется динамическое наблюдение.
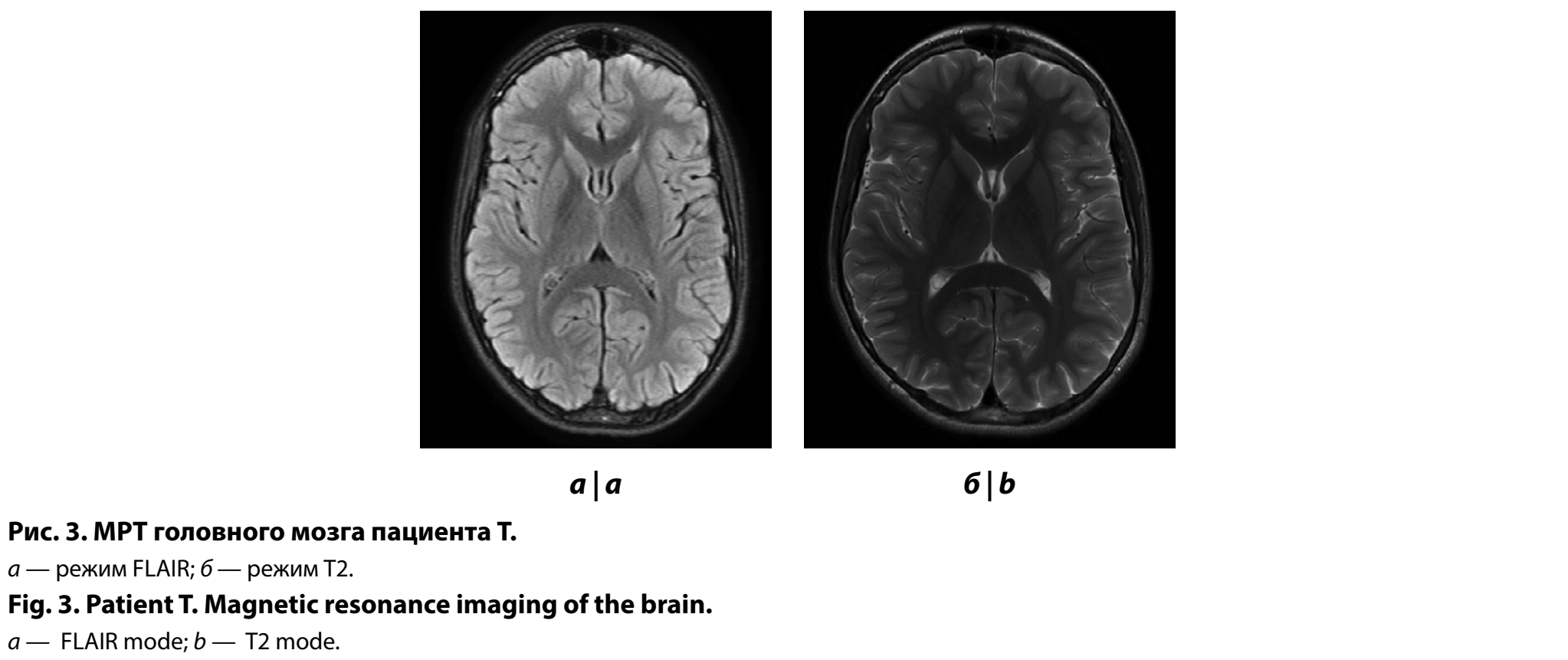
С целью оценки структур головного мозга проведено МРТ головного мозга — данных за объёмное/очаговое поражение вещества головного мозга не получено (рис. 3).
Ребёнок наблюдается в Центре в течение 3 лет, нарастания гипертрофии миокарда или обструкции выводного тракта левого желудочка не отмечено, на фоне приёма бета-блокатора и антагониста минералокортикоидных рецепторов нормализовалась ЧСС и улучшилась диастолическая функция, уровень NTproBNP в пределах референса (до 35 пг/мл). В анализах крови сохраняется повышенный уровень лактата (4,6–6,3 ммоль/л), несмотря на проводимую метаболическую терапию (левокарнитин, убидекаренон, тиамин, инозин + никотинамид + рибофлавин + янтарная кислота — курсовые приёмы). Учитывая отсутствие эпилептических приступов, от инициации противосудорожной терапии решено воздержаться. Также, принимая во внимание особенности основного заболевания, мальчик наблюдается офтальмологом, данных за атрофию зрительных нервов нет, отмечаются фоновая ретинопатия и ретинальные сосудистые изменения.
Обсуждение
Митохондриальные заболевания характеризуются мультисистемным поражением, вариабельностью клинической картины и часто летальными исходами в раннем возрасте. Пациенты с ДОФ-10 нередко умирают до 1 года, у них отмечается задержка физического и умственного развития, миопатия, снижение когнитивных способностей и поражение сердечно-сосудистой системы [12].
С. Zhou и соавт. в 2022 г. опубликовали анализ наблюдения 42 случаев ДОФ-10 у детей [13]. У всех пациентов отмечался лактат-ацидоз, часто встречался повышенный уровень аланина (53%), у 35 (83%) детей — поражение сердечно-сосудистой системы, у 31 (74%) — задержка развития, у 28 (67%) — гипотония, у 27 (64%) — умственная отсталость, у 25 (59%) — задержка психомоторного развития, 16 (38%) детей страдали от атрофии зрительного нерва. Неврологическая симптоматика у 20 (47%) пациентов была представлена в виде энцефалопатии, у 15 (36%) — судорог, у 7 (17%) — атаксии, у 5 (12%) — когнитивными нарушениями и нарушениями речи, у 3 (7%) — спастичностью. Поражение сердечно-сосудистой системы выявлено у 35 (83%) детей, в 32 случаях — гипертрофия миокарда, в единичных случаях — нарушения ритма и дефект межпредсердной перегородки. При анализе исходов авторами отмечено, что 15 (36%) пациентов умерли в младенчестве, а 1 пациент скончался в 23 года; 25 детей дожили до подросткового возраста с низким качеством жизни. У нашего пациента не отмечалось выраженной неврологической симптоматики, ему не требовалось назначение противосудорожной терапии, однако вовлечённость сердечно-сосудистой системы являлась показанием для назначения препаратов, улучшающих диастолическую функцию, контролирующих ритм и профилактирующих прогрессирование фиброзных изменений.
Анализ данных молекулярно-генетической диагностики показал, что 39% мутаций локализовались в 8-м экзоне, что позволяет предположить его функциональную значимость, а наиболее неблагоприятное течение заболевания отмечалось при выявлении вариантов c.1282G>A и c.1232C>T [13]. В нашем наблюдении один из нуклеотидных вариантов c.1430G>A был также локализован в 8-м экзоне гена MTO1 и был описан в научной литературе в качестве причины заболевания ещё у 4 пациентов (но в сочетании с иным вторым нуклеотидным вариантом), при этом у 3 детей дебют отмечался с рождения, а летальный исход зарегистрирован до 3 лет. Наиболее благоприятное течение заболевания отмечено при варианте c.1390C>T, при котором возраст пациентов на момент публикации материалов составлял 12–30 лет.
Патогенетическая терапия митохондриальных заболеваний в настоящее время не разработана. При ДОФ-10 рекомендовано поддерживающее метаболическое лечение [14], потенциально позитивное действие могут оказать убидекаренон, левокарнитин, дихлорацетат, витамин С, рибофлавин, тиамин и витамин Е.
Заключение
Несмотря на то что подавляющее большинство митохондриальных заболеваний, включая ДОФ-10, не имеет специфического лечения, диагностика данных случаев в клинической практике крайне важна для верной маршрутизации пациента и выбора оптимальной команды специалистов для оказания мультидисциплинарной медицинской помощи, а также с целью семейного консультирования родителей, которые являются носителями патогенных вариантов, по вопросам дальнейшего деторождения.
1. Wallace D.C. Mitochondrial diseases in man and mouse. Science. 1999; 283(5407): 1482–8. https://doi.org/10.1126/science.283.5407.1482
2. Schlieben L.D., Prokisch H. The dimensions of primary mitochondrial disorders. Front. Cell Dev. Biol. 2020; 8: 600079. https://doi.org/10.3389/fcell.2020.600079
3. Li R., Li X., Yan Q., Qin Mo J., Guan M.X. Identification and characterization of mouse MTO1 gene related to mitochondrial tRNA modification. Biochim. Biophys. Acta. 2003; 1629(1-3): 53–9. https://doi.org/10.1016/s0167-4781(03)00160-x
4. Powell C.A., Nicholls T.J., Minczuk M. Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front. Genet. 2015; 6: 79. https://doi.org/10.3389/fgene.2015.00079
5. Tischner C., Hofer A., Wulff V., Stepek J., Dumitru I., Becker L., et al. MTO1 mediates tissue specificity of OXPHOS defects via tRNA modification and translation optimization, which can be bypassed by dietary intervention. Hum. Mol. Genet. 2015; 24(8): 2247–66. https://doi.org/10.1093/hmg/ddu743
6. O’Byrne J.J., Tarailo-Graovac M., Ghani A., Champion M., Deshpande C., Dursun A., et al. The genotypic and phenotypic spectrum of MTO1 deficiency. Mol. Genet. Metab. 2018; 123(1): 28–42. https://doi.org/10.1016/j.ymgme.2017.11.003
7. O’Byrne J.J., Tarailo-Graovac M., Ghani A., Champion M., Deshpande C., Dursun A., et al. The genotypic and phenotypic spectrum of MTO1 deficiency. Mol. Genet. Metab. 2018; 123(1): 28–42. https://doi.org/10.1016/j.ymgme.2017.11.003
8. Савостьянов К.В. Современные алгоритмы генетической диагностики редких наследственных болезней у российский пациентов. М.; 2022. https://elibrary.ru/rduzgh
9. Гандаева Л.А., Басаргина Е.Н. Гипертрофическая кардиомиопатия в структуре инфильтративных заболеваний у детей. Российский педиатрический журнал. 2023; 26(3): 152–8. https://doi.org/10.46563/1560-9561-2023-26-3-152-158 https://elibrary.ru/wvosnj
10. Гандаева Л.А., Басаргина Е.Н., Кондакова О.Б., Каверина В.Г., Пушков А.А., Жарова О.П. и др. Новый нуклеотидный вариант в гене ELAC2 у ребенка раннего возраста с гипертрофией миокарда желудочков. Российский вестник перинатологии и педиатрии. 2022; 67(4): 120–6. https://doi.org/10.21508/1027-4065-2022-67-4-120-126 https://elibrary.ru/tpigml
11. Гандаева Л.А., Басаргина Е.Н., Давыдова Ю.И., Бурыкина Ю.С., Сильнова И.В., Пушков А.А. и др. Гипертрофическая кардиомиопатия и лактат-ацидоз у ребёнка с дефицитом ацил-КоА-дегидрогеназы-9: обзор литературы и клиническое наблюдение. Неврологический журнал имени Л.О. Бадаляна. 2023; 4(4): 215–25. https://doi.org/10.46563/2686-8997-2023-4-4-215-225 https://elibrary.ru/narqpb
12. Ghezzi D., Baruffini E., Haack T.B., Invernizzi F., Melchionda L., Dallabona C., et al. Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet. 2012; 90(6): 1079–87. https://doi.org/10.1016/j.ajhg.2012.04.011
13. Zhou C., Wang J., Zhang Q., Yang Q., Yi S., Shen Y., et al. Clinical and genetic analysis of combined oxidative phosphorylation defificiency-10 caused by MTO1 mutation. Clin. Chim. Acta. 2022; 526: 74–80. https://doi.org/10.1016/j.cca.2021.12.025
14. Baruffini E., Dallabona C., Invernizzi F., Yarham J.W., Melchionda L., Blakely E.L., et al. MTO1 mutations are associated with hypertrophic cardiomyopathy and lactic acidosis and cause respiratory chain deficiency in humans and yeast. Hum. Mutat. 2013; 34(11): 1501–9. https://doi.org/10.1002/humu.22393
Канд. мед. наук, ст. науч. сотр., врач детский кардиолог, врач-педиатр ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: sdvigova-natalya@yandex.ru
Канд. мед. наук, ведущ. науч. сотр., врач детский кардиолог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: dr.gandaeva@gmail.com
Канд. мед. наук, врач-невролог, ст. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: globa@nczd.ru
Канд. мед. наук, ст. науч. сотр., врач ультразвуковой диагностики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: silnova.iv@nczd.ru
Врач генетик ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: davydova.iui@nczd.ru
Канд. мед. наук, ст. науч. сотр., врач детский кардиолог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: basarginaeu@nczd.ru
Доктор биол. наук, нач. Медико-генетического центра, зав. лаб. медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: 7443333@gmail.com
Сдвигова Н.А., Гандаева Л.А., Глоба О.В., Сильнова И.В., Давыдова Ю.И., Басаргина Е.Ю., Савостьянов К.В. Дефицит окислительного фосфорилирования, 10 тип — редкая причина гипертрофического фенотипа кардиомиопатии. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):221-227. https://doi.org/10.46563/2686-8997-2025-6-4-221-227. EDN: gdebmq
Sdvigova N.A., Gandaeva L.A., Globa O.V., Silnova I.V., Davydova Yu.I., Basargina E.Yu., Savostyanov K.V. Oxidative phosphorylation deficiency type 10 is a rare cause of hypertrophic cardiomyopathy. L.O. Badalyan Neurological Journal. 2025;6(4):221-227. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-221-227. EDN: gdebmq

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















