


Содержание
Перейти к:
 Сергей Владимирович Воронин,
Полина Алексеевна Мухортова,
Александра Алексеевна Слабикова,
Виталий Владимирович Омельяновский
Сергей Владимирович Воронин,
Полина Алексеевна Мухортова,
Александра Алексеевна Слабикова,
Виталий Владимирович Омельяновский https://doi.org/10.46563/2686-8997-2025-6-4-234-246
EDN: graekm
Перейти к:
Мышечная дистрофия Дюшенна (МДД) — одно из наиболее тяжёлых и распространённых наследственных нервно-мышечных заболеваний, характеризующееся прогрессирующей дегенерацией мышечной ткани, утратой двигательных функций и ранней смертностью пациентов. Несмотря на наличие определённых терапевтических опций, исход заболевания во многом зависит от сроков постановки диагноза и начала наблюдения. Одним из наиболее перспективных направлений раннего выявления заболевания является неонатальный скрининг, основанный на определении активности креатинфосфокиназы (КФК) в образцах сухих пятен крови с последующим генетическим подтверждением диагноза. В мировой практике проведено множество пилотных проектов скрининга новорождённых на МДД с применением различных аналитических подходов, включая флуоресцентный и иммунофлуоресцентный анализ КФК для выявления пациентов группы повышенного риска, а также применение молекулярно-генетических методов для подтверждения диагноза. В данной работе представлен обзор истории развития и текущего состояния программ скрининга на МДД в разных странах мира, а также ключевые этические и психологические аспекты досимптоматической диагностики МДД.
Участие авторов:
Воронин С.В. — разработка концепции и структуры обзора, научное редактирование текста;
Мухортова А.А. — сбор и анализ данных, интерпретация результатов, написание разделов обзора;
Слабикова А.А. — разработка концепции и структуры обзора, анализ литературы по теме, написание разделов обзора;
Омельяновский В.В. — научное редактирование текста.
Все соавторы — утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Поступила 03.11.2025
Принята к печати 01.12.2025
Опубликована 31.01.2026
Воронин С.В., Мухортова П.А., Слабикова А.А., Омельяновский В.В. Обзор международных подходов к проведению скрининга на мышечную дистрофию Дюшенна, в том числе в рамках программ неонатального скрининга. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):234-246. https://doi.org/10.46563/2686-8997-2025-6-4-234-246. EDN: graekm
Voronin S.V., Mukhortova P.A., Slabikova A.A., Omelyanovskiy V.V. Review of international approaches to screening for Duchenne muscular dystrophy including neonatal screening programs. L.O. Badalyan Neurological Journal. 2025;6(4):234-246. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-234-246. EDN: graekm
Мышечная дистрофия Дюшенна (МДД) — тяжёлое Х-сцепленное наследственное заболевание, связанное с мутациями в гене DMD (гене дистрофина) и характеризующееся прогрессирующей дегенерацией мышечных волокон [1]. Дефицит или функциональная недостаточность данного белка приводит к структурной нестабильности мышечных волокон и их прогрессирующей дегенерации, ввиду чего заболевание проявляется проксимальной мышечной слабостью и быстрым прогрессированием симптомов. Первые клинические признаки появляются у пациентов в возрасте 3–5 лет: наблюдаются нарастающая мышечная слабость, снижение толерантности к нагрузкам, быстрая утомляемость, неуклюжесть, частые падения, затруднения при подъёме по лестнице. К 11 годам пациенты утрачивают способность к самостоятельному передвижению, а к 3-му десятилетию вследствие сердечно-лёгочных осложнений наступает смерть [1].
Мышечная дистрофия Беккера (МДБ) представляет собой более мягкую форму заболевания, манифестирующую в возрасте 10–20 лет. Для неё характерны медленное прогрессирование мышечной слабости с длительным сохранением подвижности; инвалидизация, как правило, развивается не ранее 40-летнего возраста [1].
Примерно у 8% женщин-носителей дефектного гена при наличии сопутствующих патологий (например, при полной или мозаичной формах синдрома Шерешевского–Тернера) также могут проявляться симптомы МДД. При этом у женщин заболевание, как правило, протекает в относительно более лёгкой форме: помимо умеренной мышечной слабости, часто выявляются сердечно-сосудистые нарушения, включая аритмии и дилатационную кардиомиопатию [1].
Своевременное выявление МДД имеет ключевое значение для организации медицинского наблюдения, раннего начала терапии и включения пациентов в клинические исследования. Наиболее распространённым биохимическим маркером заболевания является повышение активности креатинфосфокиназы (КФК) в крови. Самые высокие уровни (до 100 раз превышающие норму) КФК отмечаются на ранних стадиях заболевания (1–2 года жизни) и имеют тенденцию к снижению с возрастом и прогрессирующей потерей мышечной массы, хотя и остаются повышенными по сравнению с нормальными популяционными значениями. Это позволяет использовать этот показатель как в неонатальном скрининге (НС), так и в скрининге детей более старших возрастных групп [2, 3].
С середины 1970-х гг. в мировой практике реализован ряд пилотных проектов по НС на МДД. Первоначально применялись флуоресцентные методы определения КФК, чувствительность и специфичность которых ограничивались влиянием фоновых факторов. В дальнейшем разработка иммунофлуоресцентных технологий позволила селективно определять мышечную изоформу фермента (КФК-ММ), повысив точность тестирования. Дополнительным шагом, начиная с 1980-х гг., стало внедрение методов молекулярно-генетической диагностики, что позволило существенно снизить число ложноположительных результатов и обеспечить более надёжное подтверждение диагноза. Кроме того, с развитием технологий в скрининг стали чаще включаться дети обоих полов, что позволяет выявлять детей — носителей патологического варианта гена, а также девочек, у которых в связи с сопутствующими заболеваниями могут проявиться симптомы МДД.
Цель работы — провести обзор пилотных программ, направленных на раннее выявление МДД, включая проекты НС и исследования, проводившиеся в других популяциях, помимо новорождённых (НР).
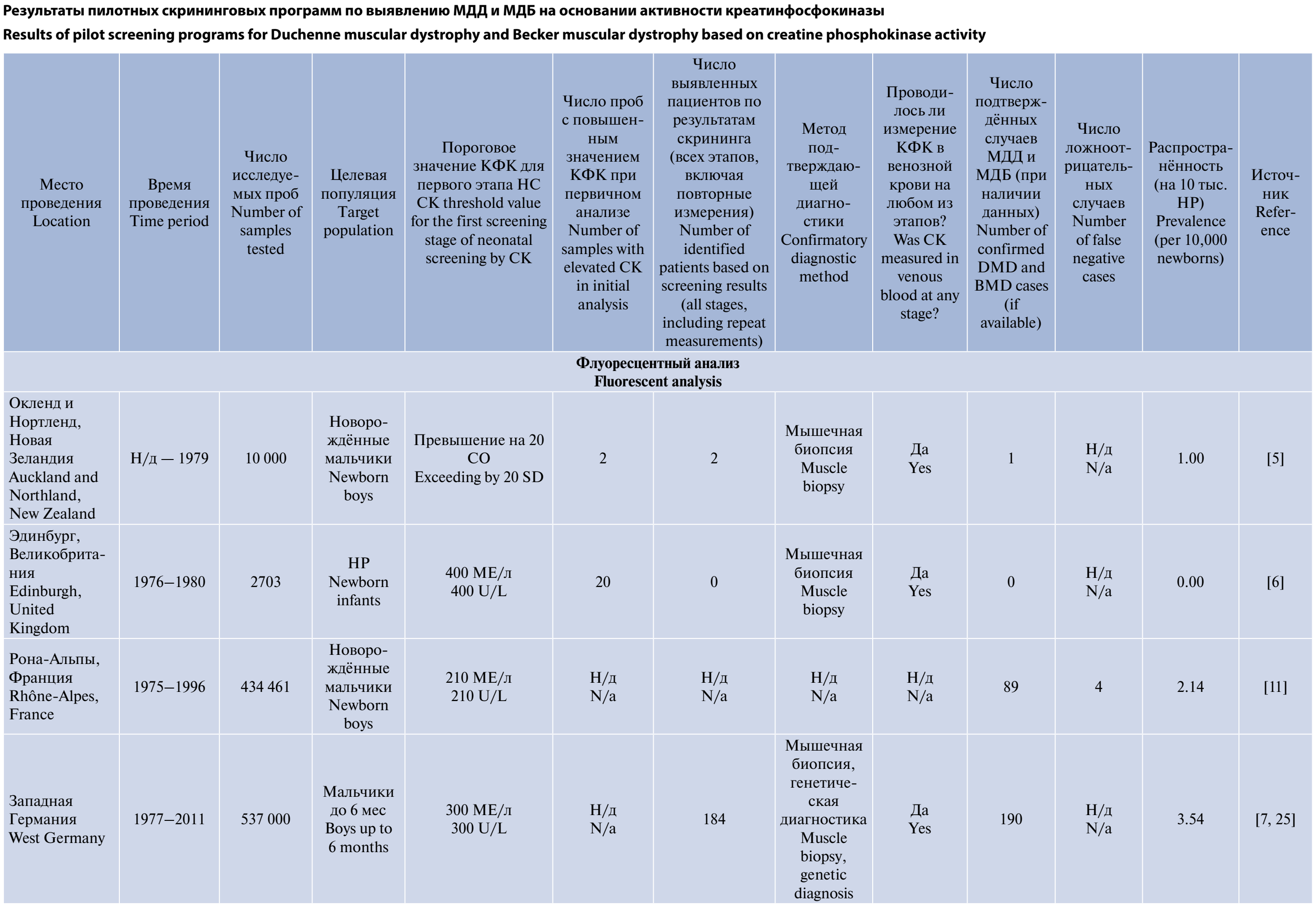
Флуоресцентный анализ
Впервые метод тестирования НР на МДД с использованием сухих пятен крови был предложен в 1975 г. Флуоресцентный анализ активности КФК был протестирован на образцах крови 1500 НР, которые уже были взяты для проведения теста на фенилкетонурию [4]. У всех, кроме 2 НР, уровень КФК соответствовал нормальным значениям, при этом результаты этих 2 НР при повторном измерении спустя несколько месяцев также соответствовали норме [2, 4]. Таким образом, несмотря на отсутствие выявленных случаев МДД, данный проект положил начало развитию скрининговых программ на МДД по всему миру.
Позже НС на МДД с использованием флуоресцентного анализа сухих пятен крови был проведён в Новой Зеландии: 10 тыс. мальчиков были обследованы на МДД с использованием образца крови на фенилкетонурию, при этом было выявлено 2 случая значительного превышения уровня КФК у детей. Проведенный впоследствии анализ крови также зафиксировал значительно повышенный уровень КФК. Биопсия мышц подтвердила диагноз у одного из детей, и у обоих младенцев уровень КФК оставался значительно повышенным. При этом согласно медицинскому осмотру отклонений от нормы у данных пациентов не выявлено [5].
В 1976–1980 гг. аналогичный пилотный проект был проведён в Эдинбурге (Великобритания). По результатам скрининга 2336 протестированных младенцев мужского пола у 16 уровень КФК превышал пороговое значение, при этом при повторном анализе данное повышение не подтвердилось ни у одного из них. С момента внедрения более чувствительных реагентов в середине 1979 г. у 850 человек, прошедших тестирование, не было обнаружено ни одного ложноположительного результата — таким образом, по результатам данного проекта не выявлено ни одного пациента с МДД [6].
В результате проведённого в 1977–1984 гг. в Западной Германии пилотного проекта, в ходе которого было обследовано 176 600 мальчиков мужского пола, у 48 из них (всем было меньше 6 мес) были значительно повышены уровни мышечной изоформы КФК — КФК-ММ (двукратное повышение более 300 МЕ/л), что авторы пилотного проекта определили как вероятное наличие диагноза МДД (частота 1:3679). Следует отметить различный возраст мальчиков на момент проведения скрининга — около 65% мальчиков, прошедших тестирование в конце 1984 г., были в возрасте 4–6 нед; 23% — от 6 нед до 6 мес, 2% — от 6 мес до года, 10% — моложе 4 нед и 0,3% — старше 1 года [7]. По результатам более поздней публикации (включающей результаты до 2011 г., когда скрининг был завершён) всего за 1977–2011 гг. было протестировано 528 410 мальчиков, в основном в возрасте 4–6 нед. За эти 34 года НС было выявлено 147 мальчиков с МДД и 33 мальчика с МДБ.
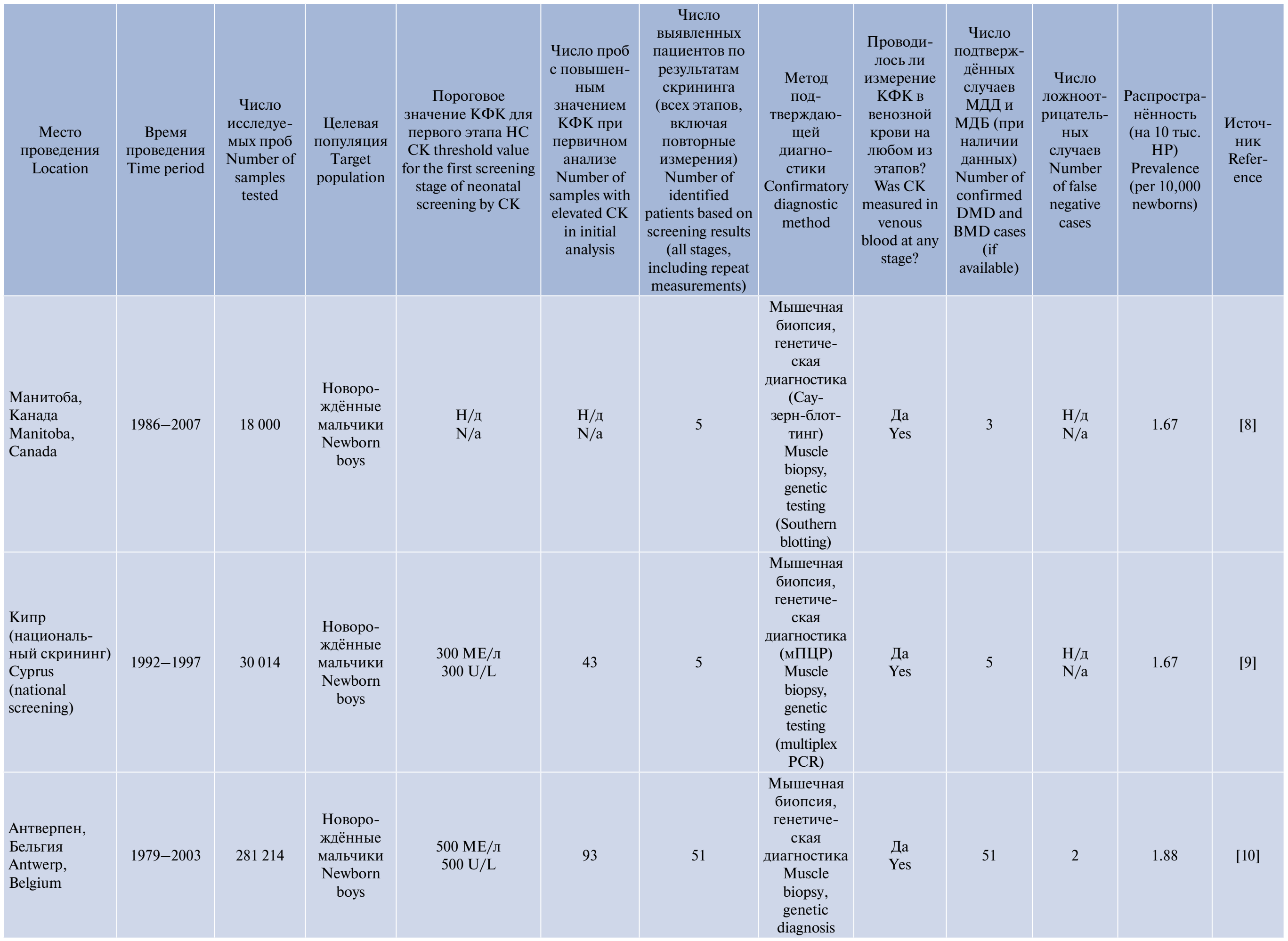
До развития методов генетической диагностики возможности проведения подтверждающей диагностики были существенно ограничены. Иногда результаты скрининга оставались неподтверждёнными до проявления клинических проявлений заболевания. Однако уже в 1988 г. были опубликованы результаты пилотного проекта в провинции Манитоба (Канада), где проводилась генетическая диагностика мутаций. По результатам скрининга из 18 тыс. новорождённых мальчиков у 3 были выявлены молекулярные делеции или дупликации [8].
Реализованный позже на Кипре национальный пилотный проект НС на МДД проводился также методом измерения уровня КФК в сухих пятнах крови с последующим генетическим подтверждением. В течение первых 6 лет программы (1992–1997 гг.) для проведения скрининга было получено 30 014 образцов, из которых 43 характеризовались изначально высокими значениями КФК (более 300 МЕ/л), однако для повторного анализа было доступно только 35 образцов. Из числа повторных проб в 30 активность КФК соответствовала нормальным значениям, а у 5 мальчиков было отмечено стойкое повышение уровня КФК. Впоследствии проведённый генетический анализ и анализ дистрофина подтвердил диагноз у всех 5 мальчиков. При этом в течение 1-го года реализации программы все образцы были проанализированы как методом фотокассет, так и количественно с помощью люминометра, после чего было отмечено, что все образцы с активностью КФК выше порогового значения (300 МЕ/л) были отобраны методом фотокассет, и в последующие годы количественный анализ с помощью люминометра проводился только тех образцов с повышенной активностью КФК, измеренной по методу фотокассет [9].
В Антверпене в 1979–2003 гг. было обследовано 281 214 новорождённых мальчиков, у 51 из которых отмечался персистирующий высокий уровень активности КФК-ММ (выше 500 МЕ/л), а у 42 мальчиков уровень КФК-ММ был повышен транзиторно и не подтверждён при повторном тестировании. Кроме того, отмечается, что 2 мальчика с МДД были пропущены при проведении скрининга [10].
Результаты пилотного проекта по НС во Франции были представлены на заседании рабочей группы Европейского нервно-мышечного центра и затем обновлены D. Bradley и E. Parsons в личной переписке c исследователями, ответственными за проведение скрининга: всего в 1975–1996 гг. было обследовано 434 461 НР, из них у 89 мальчиков были выявлены МДД/МДБ [11, 12].
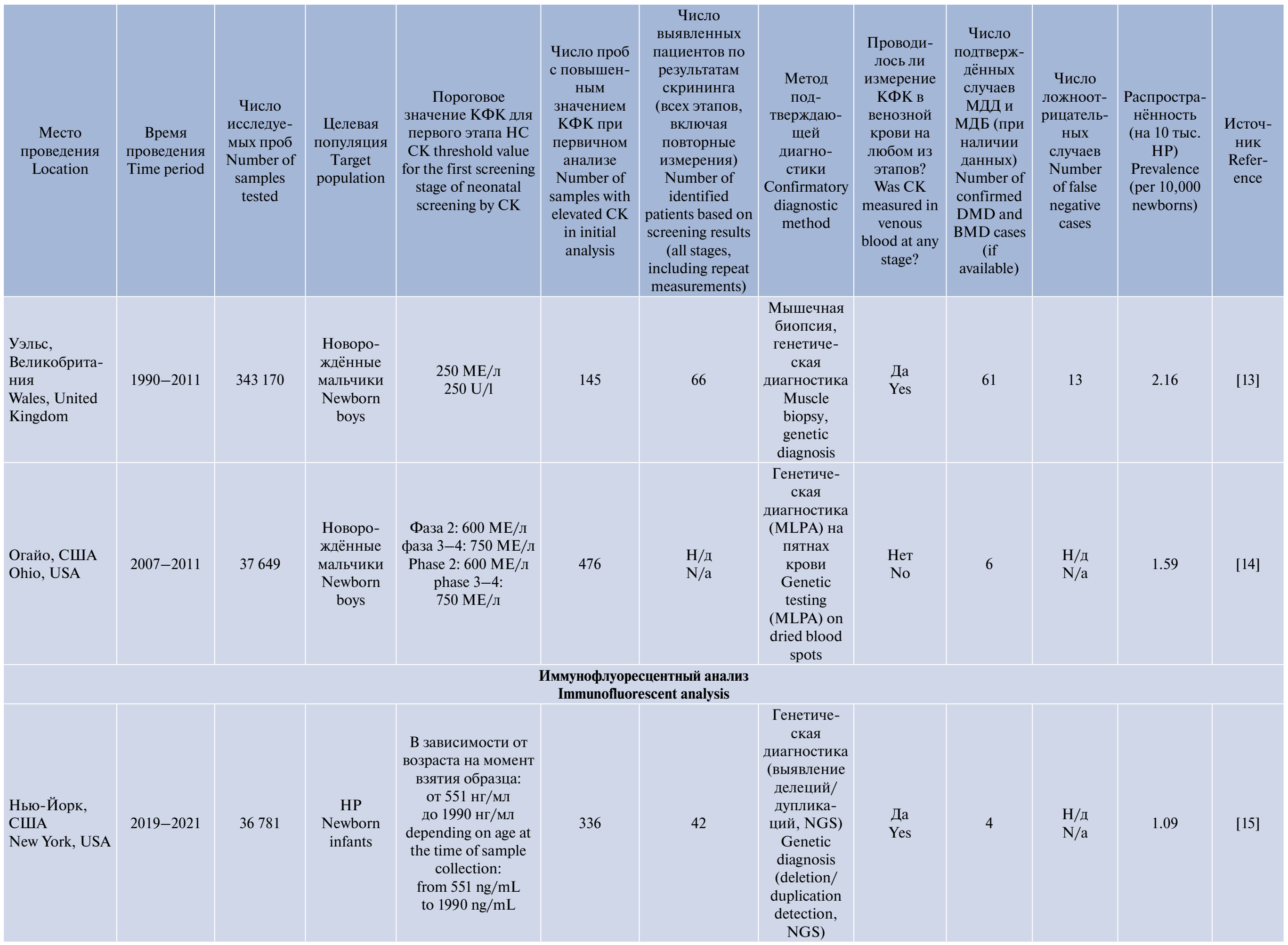
Один из самых длительных проектов скрининга был проведён в Уэльсе (с 1990 по 2011 г.). Всего в лабораторию поступило 369 780 образцов пятен крови новорождённых мальчиков, из которых у 145 образцов была выявлена повышенная активность КФК. Диагноз МДД был подтверждён у 56 пациентов, а 10 детей имели МДБ или другие дистрофии. Кроме того, у 13 детей, несмотря на отрицательный результат НС, впоследствии была выявлена МДД [13].
К 2012 г. все указанные программы, кроме скрининга в Антверпене, были прекращены. Уэльская/Антверпенская модель НС требовала повторного анализа венозной крови через несколько недель после рождения, что ограничивало её применение в странах с менее централизованной системой здравоохранения [14]. Кроме того, определение активности КФК в сухих пятнах крови методом флуоресцентного анализа ограничено влиянием эндогенного АТФ и малым объёмом элюируемого образца, что требует повышенной чувствительности метода [3].
Последний пилотный проект с применением флюорометрии (без применения иммунофлуоресцентного анализа) был проведён в штате Огайо и завершён в 2012 г. В ходе исследования мутации гена DMD (экзонные делеции) были обнаружены у 6 из 37 649 НР мужского пола (каждый из которых имел уровень КФК > 2000 ЕД/л). У 3 НР с уровнем КФК > 2000 ЕД/л аномалии гена DMD не были обнаружены, при этом были выявлены мутации генов DYSF, SGCB и FKRP, обусловливающих прочие дистрофии конечностей [14].
Иммунофлуоресцентный анализ
Следующим этапом развития скрининга на МДД стала разработка специфического теста на активность КФК-ММ в сухих пятнах крови на основе иммунофлуоресцентного анализа (ИФА). Применение данного теста позволило селективно оценивать уровень КФК-ММ в пятнах крови, что улучшило чувствительность и специфичность тестирования, а также позволило стандартизовать оценку уровня КФК среди всех регионов, организовавших скрининг.
Для оценки чувствительности и специфичности теста было проведено исследование в двух популяциях: США и Дании. Результаты скрининга на 719 архивных образцах пятен крови НР в США показали 100% чувствительность теста, в то время как в Дании по результатам тестирования были выявлены 15 из 16 образцов от мальчиков с МДД [3].
За последние 5 лет в нескольких регионах были инициированы пилотные проекты НС на МДД с использованием данного теста.
Так, в Нью-Йорке программа пилотного тестирования была запущена в октябре 2019 г. в сотрудничестве с Сетью трансляционных исследований НС (Newborn Screening Translational Research Network, NBSTRN), родительским сообществом и штатом Нью-Йорк. По результатам двухлетнего пилотного проекта был протестирован 36 781 ребёнок. Результаты скрининга разделялись на три группы: нормальные значения КФК-ММ (36 445 детей), пограничные значения КФК-ММ (296 детей) и значения КФК-ММ, предварительно соответствующие диагнозу МДД (40 детей). После повторного анализа результаты 2 детей также были отнесены в последнюю группу. По результатам генетического обследования у 4 мальчиков были обнаружены делеции или дупликации в гене DMD, соответствующие МДД или МДБ, а также выявлена 1 девочка-носительница МДД [15]. По результатам 3-летнего периода из приблизительно 650 тыс. НР у 34 детей было выявлено заболевание, и все они были направлены в специализированный центр помощи пациентам с нервно-мышечными заболеваниями [16].
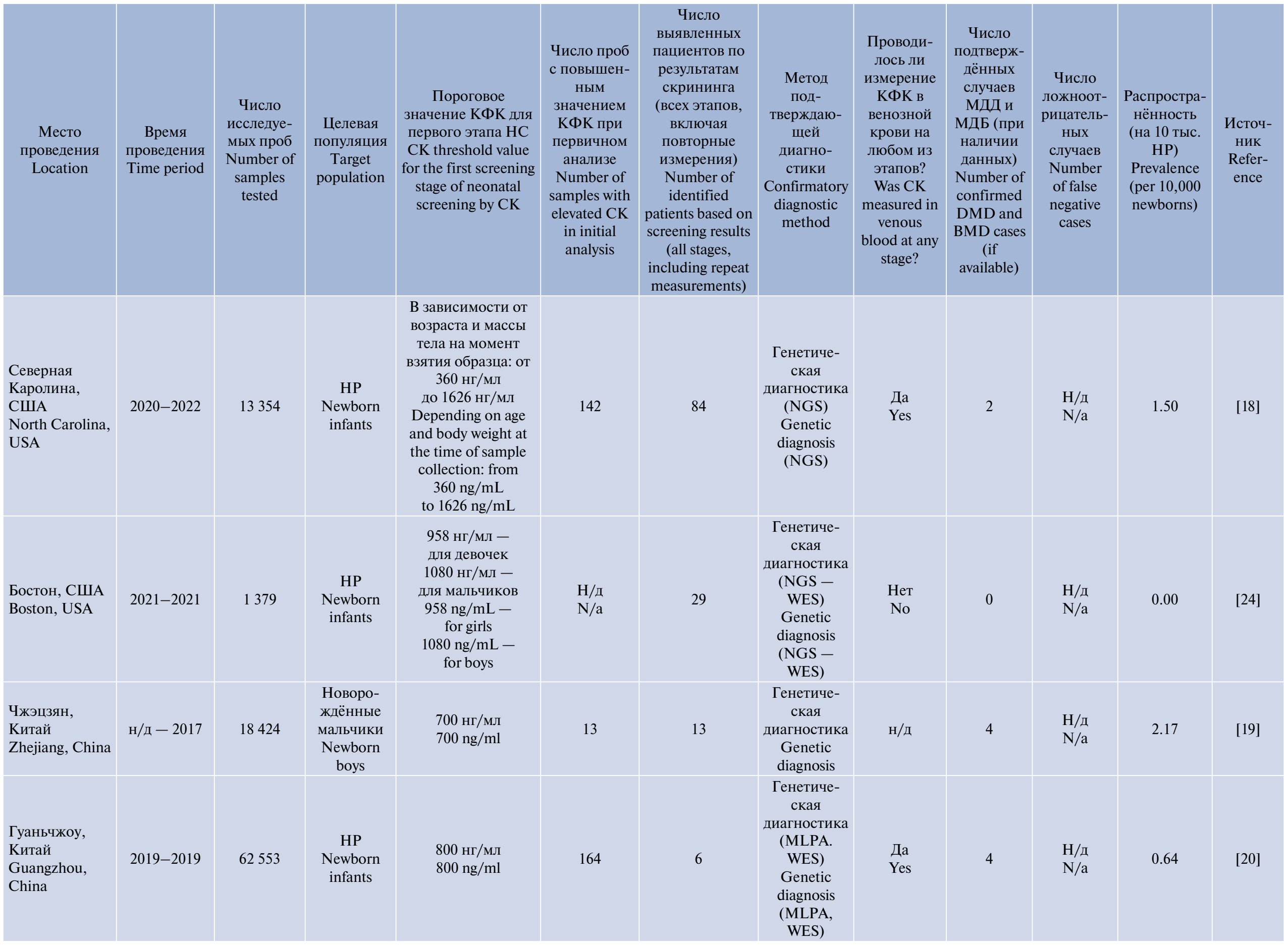
В Северной Каролине по результатам анализа 13 354 НР с помощью анализа КФК-ММ было выявлено 84 НР с повышенным уровнем КФК-ММ. По результатам генетической диагностики и измерения активности КФК в венозной крови были выявлены 2 мальчика с патогенными вариантами в гене DMD и 1 мальчик с аутосомно-доминантным патогенным вариантом в другом гене [17, 18].
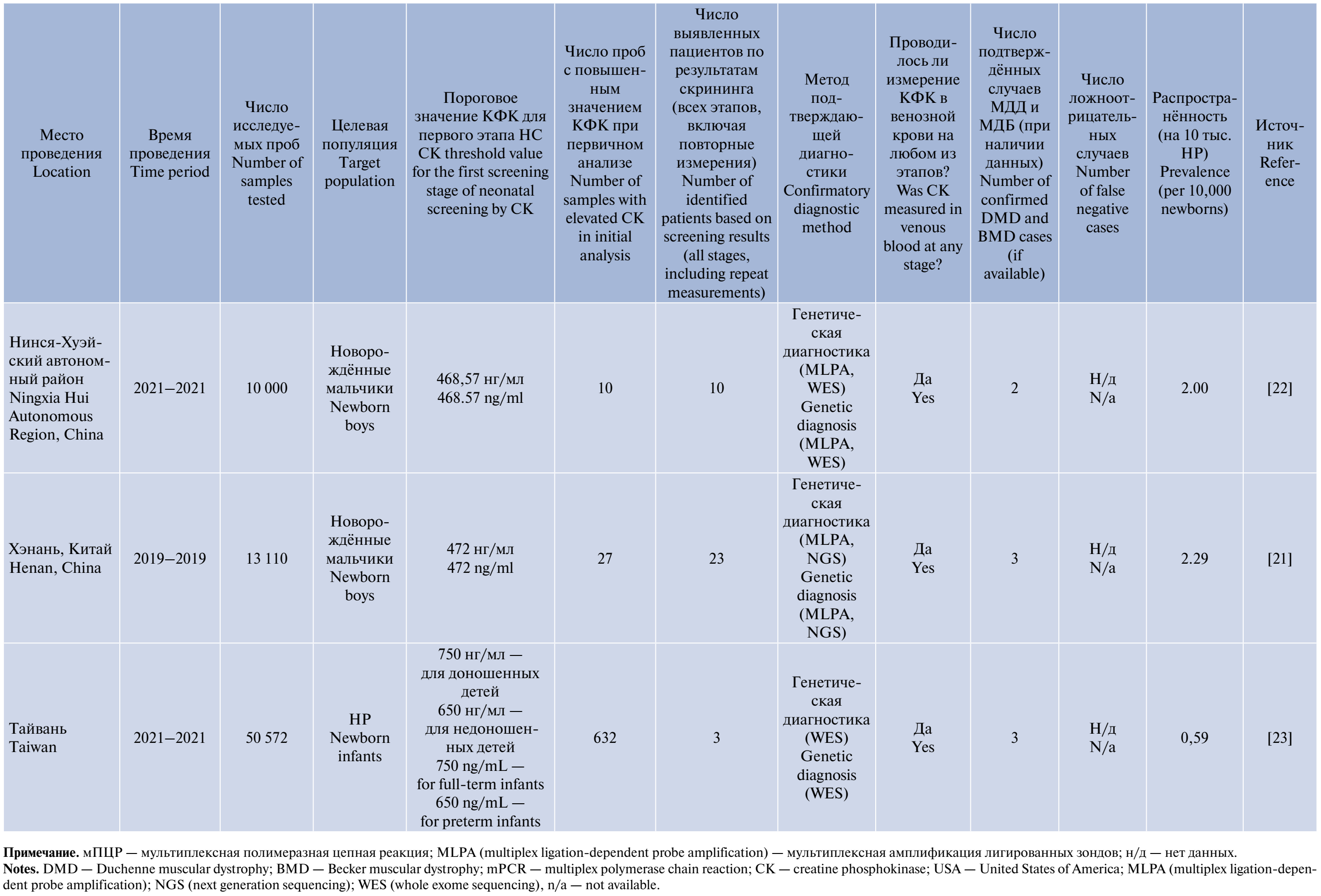
Аналогичные проекты по измерению активности КФК-ММ при помощи ИФА были инициированы в провинциях Чжэцзян, Гуанчжоу, Хэнань и Нинся-Хуэйском автономном районе в Китае.
В г. Ханчжоу (провинция Чжэцзян) согласно пилотному исследованию на 18 424 образцов высушенной крови НР было выявлено 13 образцов с повышенным значением КФК-ММ (более 700 нг/мл в крови), из которых 4 случая были классифицированы как истинно-положительные случаи МДД [19].
В Гуанчжоу в качестве порогового значения активности КФК-ММ на первом этапе скрининга устанавливалось значение 800 нг/мл. В процессе скрининга было исследовано 62 553 НР, в том числе 44 268 мальчиков и 18 285 девочек. По результатам первичного анализа высокий уровень КФК-ММ был обнаружен у 164 человек, по результатам повторного теста — у 6. Всего по результатам анализа было выявлено 4 случая МДД, ещё у 1 ребёнка найдено другое мышечное заболевание [20].
В Хэнане по результатам анализа КФК-MM у 13 110 новорождённых мальчиков было выявлено 3 случая МДД, все они имели уровень КФК-MM > 600 нг/мл [21].
По результатам пилотного проекта в Нинся-Хуэйском автономном районе, проведённом на 10 тыс. НР, было зарегистрировано 10 случаев с активностью КФК-ММ ≥ 468,57 нг/мл, из них у 2 пациентов был подтверждён диагноз МДД [22].
В Тайване измерение КФК-ММ проводили трехкратно: у 50 572 НР была проведена оценка концентрации КФК-MM, в среднем в возрасте 3 дней, повышение уровня КФК-MM (выше 750 нг/мл для доношенных детей и 650 нг/мл для недоношенных) было выявлено у 1,2% НР. По результатам двух повторных измерений были выявлены 3 пациента с повышенным уровнем КФК-ММ, при этом наличие мутаций в гене DMD было подтверждено у всех 3 пациентов [23].
Помимо улучшения предварительного этапа скрининга — определения уровня КФК в крови, совершенствуется генетическая диагностика заболевания. Так, в 2021 г. в США внедрили программу по НС МДД, предполагающую определение активности КФК c помощью вышеописанного иммунофлуоресцентного теста, последовательное таргетное секвенирование следующего поколения (targeted next-generation sequencing, tNGS) и полноэкзомное секвенирование (whole-exome sequencing, WES). Анализ tNGS/WES объединяет в себе способность обнаруживать как точечные мутации, так и делеции/дупликации гена DMD. В течение 3-месячного периода были проанализированы 1379 образцов, у 29 из которых был обнаружен повышенный уровень КФК, при этом по результатам генетического анализа ни у одного пациента МДД не была выявлена [24].
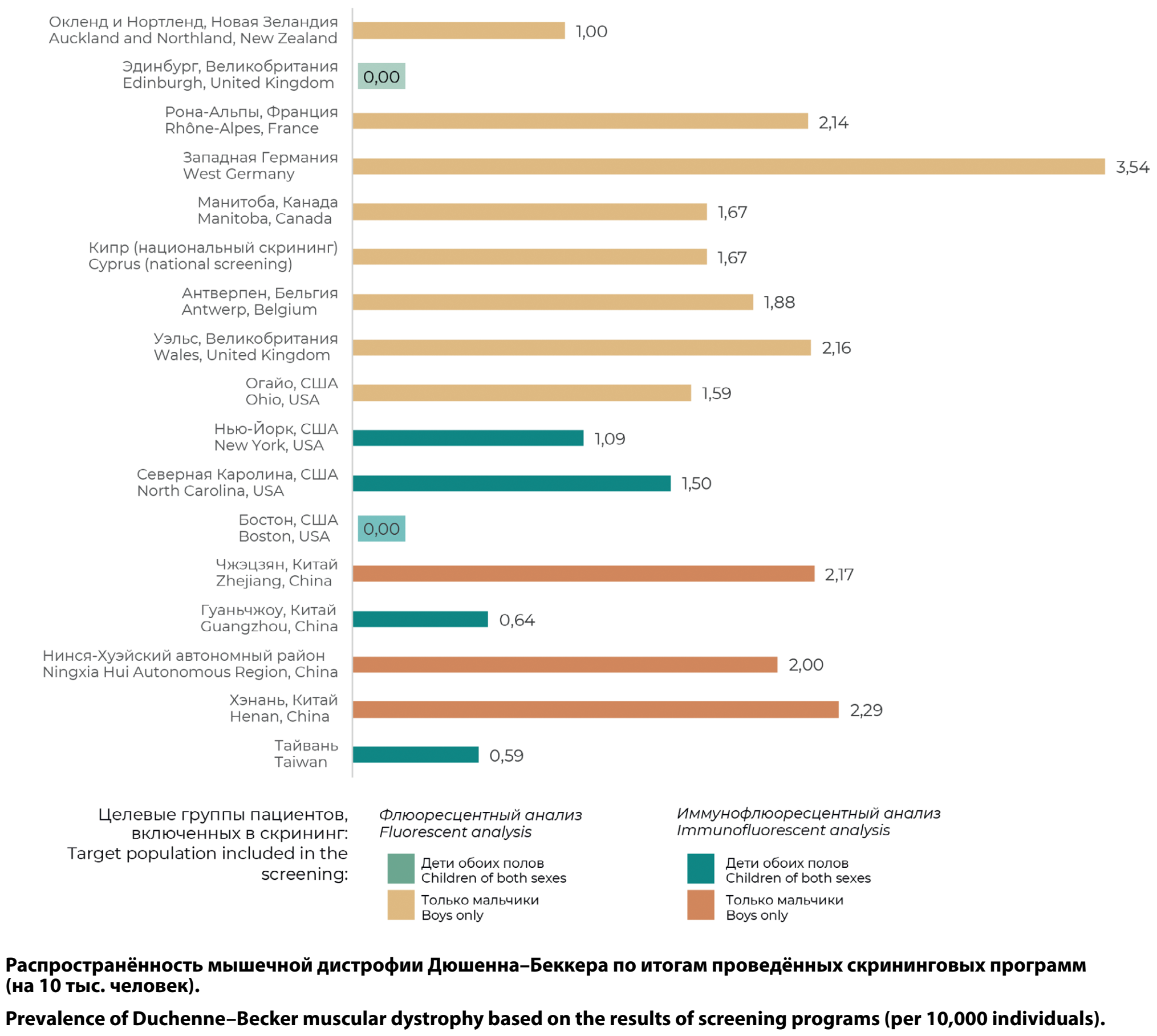
Сводная информация о всех найденных пилотных программах приведена в таблице. Распространённость МДД на 10 тыс. детей по результатам программ НС представлена на рисунке.
Прочие скрининговые программы
Помимо скрининговых программ НР для выявления МДД проводились пилотные проекты по измерению уровня КФК у детей дошкольного возраста. Так, в 1986–1987 гг. в Уэльсе было предложено тестировать мальчиков, которые не могут самостоятельно ходить к 18 мес, таким образом повысив возраст скрининга по сравнению с существовавшим в то время в Уэльсе скринингом НР. Однако скрининг прошли менее 50% неходячих мальчиков (по результатам анализа выявлено всего 2 пациента), несмотря на значительные усилия команды проекта. Организаторы пришли к выводу о том, что с учётом доли мальчиков, которые не могут ходить в 18 мес (50% от всех пациентов с МДД), и низкого охвата скринингом в ходе данного проекта могло быть выявлено только 25% мальчиков с МДД среди всей соответствующей популяции [26].
В Атланте в 2007 г. измерение уровня КФК проводили у детей, достигших возраста 1 года, в рамках плановых осмотров. Всего были проанализированы 264 образца, при этом все образцы находились в пределах нормального референсного диапазона КФК. Таким образом, дальнейшее тестирование не проводилось, и все младенцы мужского пола получили отрицательный результат скрининга [27].
В Израиле был проведён проект по выявлению мутаций в гене DMD среди населения с целью определения распространённости носительства МДД. Группа из 12 362 женщин была протестирована с использованием ряда генетических методов (MLPA с последующим секвенированием области праймера при подозрении на делецию 1 экзона). По результатам исследования у 11 женщин были обнаружены мутации в гене DMD, что соответствует частоте носительства 1:1374, при этом о семейном анамнезе заболевания сообщалось только в 3 случаях [28]. Согласно более поздним результатам анализа более 85 737 тестов на МДД частота носительства была скорректирована и составила 1:1046 женщин [29].
Этические и психологические аспекты неонатального скрининга на МДД
В процессе совершенствования технологий постепенно изменялись и этические аспекты диагностики МДД, преимущественно в контексте НС. Если до появления таргетных лекарственных препаратов, влияющих на продукцию дистрофина, ключевым оставался вопрос о целесообразности выявления заболевания до возраста, рационального для начала терапии глюкокортикостероидами, то регистрация новых терапевтических опций (аталурена в 2014 г., этеплирсена в 2016 г.) стимулировала развитие программ НС в Европе и Соединенных Штатах Америки. По результатам опроса семей, воспитывающих ребёнка с МДД (2022 г.), было показано, что 30% респондентов предпочли бы получить диагноз непосредственно после рождения пациента независимо от наличия доступных методов лечения, а подавляющее большинство (93%) выразило готовность узнать диагноз при условии существования эффективных терапевтических подходов на ранних стадиях заболевания [30]. В исследовании, проведённом в 2023 г., 80% опекунов пациентов с МДД указали, что считают установление диагноза сразу после рождения оптимальным и согласились бы на проведение НС при его доступности. Среди наиболее значимых преимуществ досимптоматической диагностики они отмечали возможность более раннего начала многопрофильного наблюдения за пациентом (58%), участие в клинических исследованиях (54%), получение психологической поддержки (56%), доступ к информации для репродуктивного планирования (54%), а также более своевременное финансовое планирование в связи с потребностями ребёнка (54%) [31].
Схожие выводы демонстрируют исследования семей с несколькими детьми, страдающими МДД: у детей младшего возраста диагноз был поставлен в среднем на 2 года раньше, чем у детей старшего возраста, причём у 27,0% из них диагноз был установлен в течение 1-го года после рождения. По мнению авторов исследования, данные результаты могут быть связаны с внедрением программ НС и сопутствующих практик — генетического консультирования, тестирования матерей на носительство мутаций и генетической диагностики братьев и сестёр [32].
В то же время остаются актуальными опасения относительно потенциального негативного влияния ранней постановки диагноза на эмоциональное развитие ребёнка и отношения в семье [33]. Однако по результатам отдельного психологического исследования, проведённого среди семей детей, получивших истинно- и ложноположительные результаты скрининга, не выявлено признаков долгосрочного нарушения отношений матери и ребёнка [34]. Более того, практически все пациенты и большинство родителей положительно относились к НС [31]. При этом родители детей, у которых диагноз был установлен в результате проведения НС, рассматривали его преимущественно как полезный опыт, тогда как семьи, в которых заболевание было диагностировано после манифестации симптомов, чаще выражали обеспокоенность, что досимптоматическая диагностика может усиливать тревожность родителей [35].
Заключение
Современное развитие технологий НС открывает возможности для выявления МДД ещё до появления клинических симптомов заболевания, что имеет критическое значение как для организации своевременного наблюдения за пациентом и начала терапии, так и для проведения генетического консультирования семей и планирования потребности пациентов на последующих этапах заболевания.
Проведённые в различных странах пилотные проекты продемонстрировали, что определение активности КФК в сухих пятнах крови с последующим подтверждением диагноза с использованием молекулярно-генетических методов может служить эффективным инструментом диагностики. Несмотря на различия в применяемых подходах, общими характеристиками программ являются многоэтапность обследования, использование специфичных тестов для оценки КФК-ММ, а также обязательное проведение генетического подтверждения.
Таким образом, международный опыт подтверждает практическую возможность включения НС на МДД в программы массового обследования НР, что приобретает особое значение в условиях появления новых терапевтических подходов, эффективность которых напрямую зависит от ранней диагностики.
1. Клинические рекомендации № 773 «Прогрессирующая мышечная дистрофия Дюшенна. Прогрессирующая мышечная дистрофия Беккера» (КР 773); 2023.
2. Dubowitz V. Screening for Duchenne muscular dystrophy. Arch. Dis. Child. 1976; 51(4): 249–51. https://doi.org/10.1136/adc.51.4.249
3. Timonen A., Lloyd-Puryear M., Hougaard D.M., Meriö L., Mäkinen P., Laitala V., et al. Duchenne muscular dystrophy newborn screening: evaluation of a new GSP® neonatal creatine kinase-MM Kit in a US and Danish population. Int. J. Neonatal. Screen. 2019; 5(3): 27. https://doi.org/10.3390/ijns5030027
4. Zellweger H., Antonik A. Newborn screening for Duchenne muscular dystrophy. Pediatrics. 1975; 55(1): 30–4.
5. Drummond L.M. Creatine phosphokinase levels in the newborn and their use in screening for Duchenne muscular dystrophy. Arch. Dis. Child. 1979; 54(5): 362–6. https://doi.org/10.1136/adc.54.5.362
6. Skinner R., Emery A.E., Scheuerbrandt G., Syme J. Feasibility of neonatal screening for Duchenne muscular dystrophy. J. Med. Genet. 1982; 19(1): 1–3. https://doi.org/10.1136/jmg.19.1.1
7. Scheuerbrandt G., Lundin A., Lövgren T., Mortier W. Screening for Duchenne muscular dystrophy: an improved screening test for creatine kinase and its application in an infant screening program. Muscle Nerve. 1986; 9(1): 11–23. https://doi.org/10.1002/mus.880090103
8. Greenberg C.R., Rohringer M., Jacobs H.K., Averill N., Nylen E., van Ommen G.J., et al. Gene studies in newborn males with Duchenne muscular dystrophy detected by neonatal screening. Lancet. 1988; 2(8608): 425–7. https://doi.org/10.1016/s0140-6736(88)90414-x
9. Drousiotou A., Ioannou P., Georgiou T., Mavrikiou E., Christopoulos G., Kyriakides T., et al. Neonatal screening for Duchenne muscular dystrophy: a novel semiquantitative application of the bioluminescence test for creatine kinase in a pilot national program in Cyprus. Genet. Test. 1998; 2(1): 55–60. https://doi.org/10.1089/gte.1998.2.55
10. Eyskens F., Philips E.G.P. Newborn screening for Duchenne muscular dystrophy. The experience in the province of Antwerp. Neuromuscul. Disord. 2006; 16(9): 721.
11. Parsons E.P., Clarke A.J., Hood K., Lycett E., Bradley D.M. Newborn screening for Duchenne muscular dystrophy: a psychosocial study. Arch. Dis. Child Fetal. Neonatal. Ed. 2002; 86(2): F91–5. https://doi.org/10.1136/fn.86.2.f91
12. van Ommen G.J., Scheuerbrandt G. Neonatal screening for muscular dystrophy. Consensus recommendation of the 14th workshop sponsored by the European Neuromuscular Center (ENMC). Neuromuscul. Disord. 1993; 3(3): 231–9. https://doi.org/10.1016/0960-8966(93)90065-r
13. Moat S.J., Bradley D.M., Salmon R., Clarke A., Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur. J. Hum. Genet. 2013; 21(10): 1049–53. https://doi.org/10.1038/ejhg.2012.301
14. Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J., et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012; 71(3): 304–13. https://doi.org/10.1002/ana.23528
15. Tavakoli N.P., Gruber D., Armstrong N., Chung W.K., Maloney B., Park S., et al. Newborn screening for Duchenne muscular dystrophy: A two-year pilot study. Ann. Clin. Transl. Neurol. 2023; 10(8): 1383–96. https://doi.org/10.1002/acn3.51829
16. Lee B.H., Deng S., Chiriboga C.A., Kay D.M., Irumudomon O., Laureta E., et al. Newborn screening for spinal muscular atrophy in New York state: clinical outcomes from the first 3 years. Neurology. 2022; 99(14): e1527–37. https://doi.org/10.1212/WNL.0000000000200986
17. Kucera K., Boyea M.B., Migliore B., Robles V., Cope H., Rehder C., et al. P497: Two years of newborn screening for Duchenne muscular dystrophy in North Carolina: Results from early check. Genet. Med. Open. 2023; 1(1 Suppl.): 100544. https://doi.org/10.1016/j.gimo.2023.100544
18. Kucera K.S., Boyea B.L., Migliore B., Potter S.N., Robles V.R., Kutsa O., et al. Two years of newborn screening for Duchenne muscular dystrophy as a part of the statewide Early Check research program in North Carolina. Genet. Med. 2024; 26(1): 101009. https://doi.org/10.1016/j.gim.2023.101009
19. Ke Q., Zhao Z.Y., Griggs R., Wiley V., Connolly A., Kwon J., et al. Newborn screening for Duchenne muscular dystrophy in China: follow-up diagnosis and subsequent treatment. World J. Pediatr. 2017; 13(3): 197–201. https://doi.org/10.1007/s12519-017-0036-3
20. Jia X., Jiang X., Huang Y. A pilot study of newborn screening for Duchenne muscular dystrophy in Guangzhou. Heliyon. 2022; 8(10): e11071. https://doi.org/10.1016/j.heliyon.2022.e11071
21. Jia C., Zhao D., Li Y., Gao Y., Zhang X., Li X., et al. Newborn screening and genomic analysis of Duchenne muscular dystrophy in Henan, China. Clin. Chim. Acta. 2023; 539: 90–6. https://doi.org/10.1016/j.cca.2022.11.024
22. Jing M., Wang Y., Jing X.Y., Mao X.M. Screening for Duchenne muscular dystrophy in newborns in the Ningxia region. Zhongguo Dang Dai Er Ke Za Zhi. 2024; 26(3): 258–61. https://doi.org/10.7499/j.issn.1008-8830.2309151 (in Chinese)
23. Chien Y.H., Lee N.C., Weng W.C., Chen L.C., Huang Y.H., Wu C.S., et al. Duchenne muscular dystrophy newborn screening: the first 50,000 newborns screened in Taiwan. Neurol. Sci. 2022; 43(7): 4563–6. https://doi.org/10.1007/s10072-022-06128-2
24. Parad R.B., Sheldon Y., Bhattacharjee A. Implementation of hospital-based supplemental Duchenne muscular dystrophy newborn screening (sDMDNBS): a pathway to broadening adoption. Int. J. Neonatal. Screen. 2021; 7(4): 77. https://doi.org/10.3390/ijns7040077
25. Ellis J.A., Vroom E., Muntoni F. 195th ENMC international workshop: Newborn screening for Duchenne muscular dystrophy 14-16th December, 2012, Naarden, The Netherlands. Neuromuscul. Disord. 2013; 23(8): 682–9. https://doi.org/10.1016/j.nmd.2013.05.008
26. Fenton-May J., Bradley D.M., Sibert J.R., Smith R., Parsons E.P., Harper P.S., et al. Screening for Duchenne muscular dystrophy. Arch. Dis. Child. 1994; 70(6): 551–2. https://doi.org/10.1136/adc.70.6.551
27. Cyrus A., Street N., Quary S., Kable J., Kenneson A., Fernhoff P. Clinic-based infant screening for Duchenne muscular dystrophy: a feasibility study. PLoS Curr. 2012; 4: e4f99c5654147a. https://doi.org/10.1371/4f99c5654147a
28. Cohen G., Shtorch-Asor A., Ben-Shachar S., Goldfarb-Yaacobi R., Kaiser M., Rosenfeld R., et al. Large scale population screening for Duchenne muscular dystrophy-Predictable and unpredictable challenges. Prenat. Diagn. 2022; 42(9): 1162–72. https://doi.org/10.1002/pd.6201
29. Singer A., Aartsma-Rus A., Grinshpun-Cohen J., Sagi-Dain L. Lessons learned from the first national population-based genetic carrier-screening program for Duchenne muscular dystrophy. Genet. Med. 2023; 25(12): 100981. https://doi.org/10.1016/j.gim.2023.100981
30. Crossnohere N.L., Armstrong N., Fischer R., Bridges J.F.P. Diagnostic experiences of Duchenne families and their preferences for newborn screening: A mixed-methods study. Am. J. Med. Genet. C Semin. Med. Genet. 2022; 190(2): 169–77. https://doi.org/10.1002/ajmg.c.31992
31. Ji C., Kariyawasam D.S., Sampaio H., Lorentzos M., Jones K.J., Farrar M.A. Newborn screening for Duchenne muscular dystrophy: the perspectives of stakeholders. Lancet Reg. Health West. Pac. 2024; 45: 101049. https://doi.org/10.1016/j.lanwpc.2024.101049
32. Bhattacharyya O., Campoamor N.B., Armstrong N., Freed M., Schrader R., Crossnohere N.L., et al. Assessing the benefits and harms associated with early diagnosis from the perspective of parents with multiple children diagnosed with Duchenne muscular dystrophy. Int. J. Neonatal. Screen. 2024; 10(2): 32. https://doi.org/10.3390/ijns10020032
33. Bowman J.E. Screening newborn infants for Duchenne muscular dystrophy. BMJ. 1993; 306(6874): 349. https://doi.org/10.1136/bmj.306.6874.349
34. Parsons E.P., Clarke A.J., Hood K., Lycett E., Bradley D.M. Newborn screening for Duchenne muscular dystrophy: a psychosocial study. Arch. Dis. Child Fetal. Neonatal. Ed. 2002; 86(2): F91–5. https://doi.org/10.1136/fn.86.2.f91
35. Chung J., Smith A.L., Hughes S.C., Niizawa G., Abdel-Hamid H.Z., Naylor E.W., et al. Twenty-year follow-up of newborn screening for patients with muscular dystrophy. Muscle Nerve. 2016; 53(4): 570–8. https://doi.org/10.1002/mus.24880
Канд. мед. наук, гл. врач, доцент кафедры биохимической генетики и наследственных болезней обмена веществ ФГБНУ «МГНЦ», Москва, Россия
e-mail: voroninsvvlad@mail.ru
Гл. спец. отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении ФГБУ «ЦЭККМП» Минздрава России, 109028, Москва, Россия
e-mail: muhortova@rosmedex.ru
Ведущий специалист отдела методологического обеспечения проведения комплексной оценки технологий в здравоохранении ФГБУ «ЦЭККМП» Минздрава России, 109028, Москва, Россия
e-mail: slabikova@rosmedex.ru
Доктор мед. наук, профессор, генеральный директор ФГБУ «ЦЭККМП» Минздрава России, 109028, Москва, Россия
e-mail: vvo@rosmedex.ru
Воронин С.В., Мухортова П.А., Слабикова А.А., Омельяновский В.В. Обзор международных подходов к проведению скрининга на мышечную дистрофию Дюшенна, в том числе в рамках программ неонатального скрининга. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):234-246. https://doi.org/10.46563/2686-8997-2025-6-4-234-246. EDN: graekm
Voronin S.V., Mukhortova P.A., Slabikova A.A., Omelyanovskiy V.V. Review of international approaches to screening for Duchenne muscular dystrophy including neonatal screening programs. L.O. Badalyan Neurological Journal. 2025;6(4):234-246. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-234-246. EDN: graekm

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















