


Содержание
Перейти к:
 Юлия Сергеевна Бурыкина,
Наталия Андреевна Сдвигова,
Елена Николаевна Басаргина,
Оксана Валерьевна Глоба,
Алексей Львович Куренков,
Илья Сергеевич Жанин,
Александр Алексеевич Пушков,
Кирилл Викторович Савостьянов
Юлия Сергеевна Бурыкина,
Наталия Андреевна Сдвигова,
Елена Николаевна Басаргина,
Оксана Валерьевна Глоба,
Алексей Львович Куренков,
Илья Сергеевич Жанин,
Александр Алексеевич Пушков,
Кирилл Викторович Савостьянов https://doi.org/10.46563/2686-8997-2025-6-4-195-208
EDN: mhpsyv
Перейти к:
Введение. Дилатационный фенотип кардиомиопатии (ДКМП) представляет собой группу заболеваний миокарда, основными характеристиками которых являются расширение полости левого желудочка и нарушение его сократительной функции. Благодаря значительному прогрессу в области молекулярной генетики в настоящее время идентифицировано свыше 50 генов, обусловливающих развитие первичной формы КМП, причём на долю мутаций гена MYH7 приходится до 50% генетически подтверждённых случаев. Цель исследования — установить клинические и генетические характеристики КМП с дилатационным фенотипом, обусловленной мутациями в гене MYH7, у детей.
Материалы и методы. В работе проанализированы данные 250 детей с дилатационным фенотипом КМП, обследованных в кардиологическом отделении ФГАУ «НМИЦ здоровья детей» Минздрава России в 2015–2024 гг. Использованы современные методы молекулярно-генетической диагностики и комплексное клинико-инструментальное обследование с оценкой динамики состояния в течение 5 лет.
Результаты. Изолированная MYH7-обусловленная КМП с дилатационным фенотипом подтверждена у 80 (40,8%) неродственных детей с выявленными каузальными вариантами. Клиническими характеристиками заболевания являются ранний дебют (более 80% случаев на 1-м году жизни), частое сочетание с некомпактным миокардом (85%) и развитие обратного ремоделирования миокарда левого желудочка на фоне лечения. Установлено, что варианты в экзонах 19–23 ассоциированы с тяжёлой митральной недостаточностью, требующей хирургического вмешательства в раннем возрасте. В 3 случаях отмечено редкое сочетание КМП и миопатии.
Заключение. Исследование подтверждает важную роль вариантов гена MYH7 в развитии КМП у детей. MYH7-обусловленные формы заболевания при своевременно начатой и адекватной медикаментозной терапии характеризуются благоприятным прогнозом, о чём свидетельствует высокая 5-летняя выживаемость (97,4%) пациентов. Проведение молекулярно-генетической диагностики имеет важное значение в верификации диагноза, организации раннего обследования родственников, прогнозировании течения заболевания. Систематизация клинико-генетических характеристик создаёт фундамент для разработки персонализированных подходов к лечению. Междисциплинарный подход к ведению пациентов с КМП и экстракардиальными проявлениями приобретает особое значение.
Соблюдение этических стандартов. Исследование проведено в соответствии с этическими нормами Хельсинкской декларации 1975 г. и одобрено локальным этическим комитетом ФГАУ «НМИЦ здоровья детей» Минздрава России (протокол № 10 от 28.08.2020). Все участники или их законные представители предоставили информированное согласие на участие.
Участие авторов:
Бурыкина Ю.С. — концепция и дизайн исследования, статистическая обработка данных, написание и редактирование текста;
Сдвигова Н.А. — редактирование текста;
Басаргина Е.Н. — редактирование текста;
Глоба О.В. — сбор и обработка материала, редактирование текста;
Куренков А.Л. — сбор и обработка материала, редактирование текста;
Жанин И.С. — редактирование текста;
Пушков А.А. — сбор и обработка материала, редактирование текста;
Савостьянов К.В. — концепция и дизайн исследования, редактирование текста.
Все соавторы — утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Конфликт интересов. Авторы статьи заявили об отсутствии конфликта интересов.
Благодарность. Авторы благодарны семьям пациентов за поддержку нашего исследования. Авторы выражают благодарность директору ФГАУ «НМИЦ здоровья детей» Минздрава России доктору медицинских наук, профессору А.П. Фисенко за поддержку и техническую помощь в осуществлении данной работы. Авторы благодарят весь коллектив ФГАУ «НМИЦ здоровья детей» Минздрава России за возможность междисциплинарного подхода к ведению пациентов.
Поступила 08.10.2025
Принята к печати 01.12.2025
Опубликована 31.01.2026
Бурыкина Ю.С., Сдвигова Н.А., Басаргина Е.Н., Глоба О.В., Куренков А.Л., Жанин И.С., Пушков А.А., Савостьянов К.В. Одноцентровое исследование клинико-генетических характеристик дилатационного фенотипа кардиомиопатии, обусловленной вариантами в гене MYH7, у 80 неродственных детей. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):195-208. https://doi.org/10.46563/2686-8997-2025-6-4-195-208. EDN: mhpsyv
Burykina Yu.S., Sdvigova N.A., Basargina E.N., Globa O.V., Kurenkov A.L., Zhanin I.S., Pushkov A.A., Savostyanov K.V. A single-center study of the clinical and genetic characteristics of the dilated cardiomyopathy caused by MYH7 gene variants in eighty unrelated children. L.O. Badalyan Neurological Journal. 2025;6(4):195-208. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-195-208. EDN: mhpsyv
Введение
Дилатационный фенотип кардиомиопатии (ДКМП) относится к числу наиболее распространённых, составляя, по разным данным, до 60% случаев КМП [1–3]. Заболевание характеризуется прогрессирующим расширением полости левого желудочка (ЛЖ) и ухудшением его сократительной функции при отсутствии вторичных причин ремоделирования, таких как ишемия, гипертензия, метаболические расстройства и др. [4–6].
Манифестация ДКМП возможна в любом возрасте, однако наиболее часто заболевание выявляется у детей 1-го года жизни [3,7]. ДКМП отличается неблагоприятным прогнозом: примерно 40% пациентов в течение 5 лет с момента установления диагноза достигают терминальной точки наблюдения — трансплантации сердца или летального исхода [3, 7].
ДКМП имеет разнообразное происхождение. Различают первичную (генетически обусловленную) форму, преобладающую у детей, и вторичную, связанную с ишемией, воспалением, аритмиями или генетическими синдромами [3, 8–10]. Благодаря развитию методов высокопроизводительного секвенирования, всё чаще удаётся выявлять наследственные причины заболевания. В настоящее время известно более 50 генов, мутации в которых приводят к развитию ДКМП [3, 10].
Среди генетических причин ДКМП у детей наиболее часто встречаются мутации саркомерных генов, включая TPM1, TNNT2, TTN, MYH7, ACTC1, ACTN2, NEXN и др. [6, 10–12]. Ген MYH7, ответственный за синтез β-тяжёлой цепи миозина, заслуживает особого внимания — патогенные варианты этого гена обусловливают 10–50% случаев ДКМП и некомпактного миокарда [10–13].
У пациентов с мутациями в гене MYH7 нередко наблюдается сочетанное поражение сердечной и скелетной мускулатуры, проявляющееся комбинацией ДКМП и миопатии [14–18]. Заболевание у этих пациентов характеризуется более тяжёлым течением с ранним развитием сердечной недостаточности, выраженной мышечной слабостью и задержкой моторного развития [18]. Особенностью MYH7-обусловленных миопатий является преимущественное вовлечение проксимальных групп мышц, при этом гистологическое исследование часто выявляет характерные изменения по типу миозиновой миопатии [15]. Данный фенотип подчёркивает ключевую роль β-тяжёлой цепи миозина в функционировании как сердечной, так и скелетной мускулатуры, что требует междисциплинарного подхода к диагностике и лечению таких пациентов.
Существующие исследования генетических причин ДКМП, проводившиеся как в России, так и за рубежом, преимущественно охватывали взрослых пациентов [8, 11, 19, 20], тогда как аналогичные исследования в педиатрической практике практически отсутствуют.
Цель работы — оценить клинические особенности течения заболевания MYH7-обусловленной КМП на большой выборке пациентов детского возраста.
Материалы и методы
В период с 2015 по 2024 г. в кардиологическом отделении Центра обследовано 250 детей в возрасте от 0 до 17 лет 11 мес 30 дней с ДКМП. Комплексное молекулярно-генетическое тестирование позволило верифицировать первичный генез заболевания у 196 (78,4%) детей. У 54 пациентов молекулярно-генетическое исследование не выявило мутаций, ответственных за развитие КМП, а инструментальное дообследование верифицировало вторичный характер заболевания (ишемические/поствоспалительные изменения, аритмии или генетические синдромы), что позволило исключить их из последующего анализа.
В исследование включено 196 пациентов, критериями включения было наличие эхокардиографических признаков ДКМП: дилатации ЛЖ с конечно-диастолическим размером (КДР) ≥ +2 z-score и снижения сократительной функции с фракцией выброса (ФВ < 55% по Simpson) при первичной диагностике или в дебюте заболевания, первичный характер заболевания, подтверждённый генетически.
Все пациенты прошли комплексное клинико-инструментальное обследование, включавшее детальный анализ семейного анамнеза с акцентом на случаи КМП и летальных исходов среди родственников до 35 лет. Обязательный диагностический алгоритм включал физикальное обследование сердечно-сосудистой системы с оценкой функционального класса (ФК) хронической сердечной недостаточности (ХСН) — у детей младшего возраста в соответствии с классификацией Ross, у детей старше 6 лет — согласно критериям NYHA; стандартную эхокардиографию (ЭхоКГ), регистрацию электрокардиограммы (ЭКГ) в покое, суточное мониторирование ЭКГ (СМ-ЭКГ), определение уровня N-концевого фрагмента мозгового натрийуретического пептида (NTproBNP) в сыворотке крови. По показаниям проводили магнитно-резонансную томографию (МРТ) сердца. Некомпактный миокард определяли при двуслойной структуре миокарда ЛЖ с соотношением толщины некомпактного слоя к компактному от 2:1 и более с глубокими межтрабекулярными лакунами. В случае соотношения слоев менее 2 диагностировали повышенную трабекулярность.
Для всех пациентов проводили индивидуальный подбор медикаментозной терапии ХСН в соответствии с клиническими рекомендациями: ингибитор ангиотензинпревращающего фермента или антагонист рецепторов ангиотензина 2, β-адреноблокатор, антагонист минералокортикоидных рецепторов, по показаниям — диуретик, сердечный гликозид, профилактика тромбообразования. Динамическое наблюдение с повторной оценкой клинико-лабораторных показателей проводили через 1 год и 5 лет после первичного обследования.
Молекулярно-генетическое исследование проводили всем пациентам с использованием технологии высокопроизводительного секвенирования для анализа таргетных областей генома, включающих 404 гена с использованием панели, разработанной в лаборатории медицинской геномики Медико-генетического центра ФГАУ «НМИЦ здоровья детей» Минздрава России [4]. В случае, если специфическая панель не выявляла причину заболевания, у пациентов были исследованы таргетные области клинического или полного экзомов. У пациентов брали 1–2 мл цельной венозной крови в пробирку-вакутейнер с антикоагулянтом ЭДТА. Из образцов крови выделяли геномную ДНК с использованием набора реактивов «DNA Blood Mini Kit» («Qiagen») на автоматической станции «QIAQUBE» («Qiagen»). После оценки качества и количества ДНК на флоуриметре «Qubit 3.0» («Invitrogen») пробоподготовку проводили при помощи набора реагентов «KAPA HyperPlusKit» («Roche»). Секвенирование осуществляли на платформе «NextSeq» («Illumina», 300 циклов, парноконцевые чтения). Минорные варианты генов с частотой встречаемости менее 0,5% для рецессивных заболеваний и менее 0,01% для доминантных заболеваний (согласно базе данных Exome Aggregation Consortium) подвергали биоинформатическому анализу с использованием программного обеспечения «Alamut Batch» и «Alamut Focus» («Interactive Biosoftware»). Для валидации результатов и проведения анализа семейной сегрегации использовали технологию двунаправленного секвенирования по Сэнгеру. Ранее неописанные варианты анализировали с помощью программы «Alamut Visual» («Interactive Biosoftware») и российского «Руководства по интерпретации последовательностей ДНК человека» [21]. Клиническую значимость описанных генетических вариантов оценивали на основе базы данных мутаций человека «HGMD Professional» [22].
Результаты
Генетический анализ материала 196 детей с ДКМП позволил идентифицировать 203 генетических варианта различной клинической значимости. Наибольшая доля генетических вариантов (40,89%; n = 83) была обнаружена в гене MYH7. При этом 3 случая представляли собой комбинации с другими патогенными вариантами (в генах ACTC1, SCN5A и биаллельной мутацией ABCC6, обусловливающей развитие артериальной кальцификации [23]), что потребовало их отдельного анализа от пациентов с изолированными MYH7 вариантами (n = 80). Все варианты были гетерозиготными.
Значимых отличий по полу в исследуемой группе не выявлено: 42 мальчика и 38 девочек (табл. 1). Изменения со стороны сердца были обнаружены во время пренатального скрининга у 12 пациентов, медиана возраста выявления у остальных пациентов составила 2 мес. У 21 ребёнка изменения со стороны сердца впервые были отмечены на фоне интеркуррентной инфекции, у остальных — в рамках профилактических осмотров.
Основными клиническими проявлениями, послужившими поводом для обращения за медицинской помощью, стали повышенная утомляемость и снижение переносимости нагрузок (61,3%), одышка (62,5%), отставание в физическом развитии (13,8%), потеря сознания и гастроинтестинальные симптомы (3,8%). В дебюте заболевания преобладал II ФК сердечной недостаточности.
Семейный характер заболевания подтверждён у 31 (38,8%) пациента. В 18 семьях КМП у родителей была диагностирована при каскадном скрининге после выявления патологии у ребёнка. В 9 случаях ребёнок обследовался целенаправленно в связи с установленным семейным характером заболевания. У 4 пациентов родители с подтверждённым диагнозом КМП отказались от генетического тестирования. Во всех наблюдаемых случаях фенотипические проявления ремоделирования миокарда у родителей и детей были идентичны. У 12 (15%) детей был идентифицирован каузальный вариант de novo.
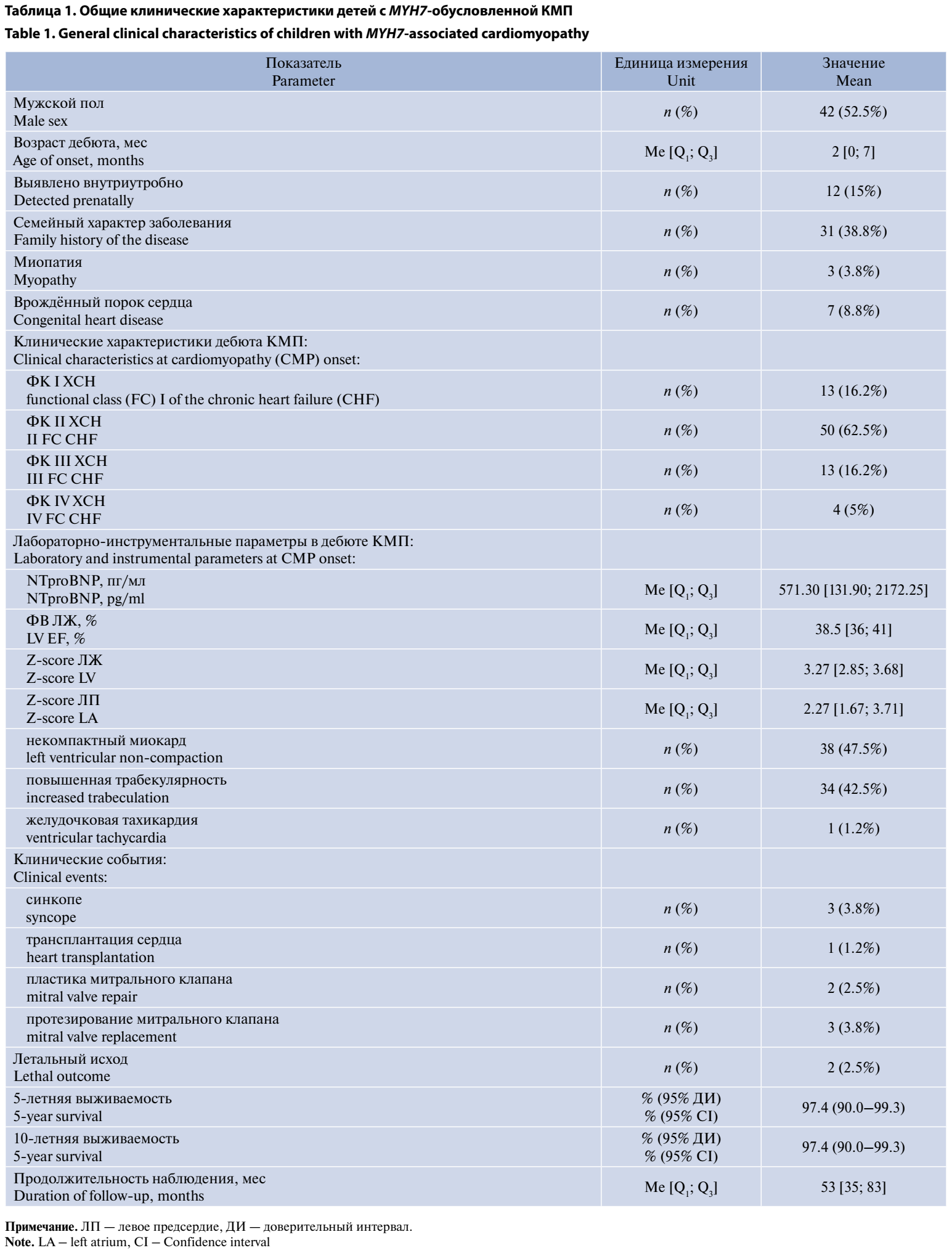
В качестве основного биомаркера тяжести ХСН использовали уровень NTproBNP. На фоне индивидуально подобранной медикаментозной терапии при среднем сроке наблюдения 53 мес было зафиксировано снижение данного показателя (рис. 1): с исходного уровня 571,30 пг/мл до 186,05 пг/мл [96,19; 416,40] через 1 год наблюдения и 115,0 пг/мл [60,8; 166,0] через 5 лет наблюдения.
При первичном ЭхоКГ-обследовании у всех пациентов были выявлены характерные признаки дилатационного ремоделирования миокарда, причём более чем у 80% детей с MYH7-обусловленной КМП отмечались признаки некомпактного миокарда или повышенной трабекулярности. При динамическом наблюдении зафиксирована умеренная положительная динамика показателей (рис. 1): z-score КДР ЛЖ нормализовался с +3,27 до +1,97 через 1 год и до +1,42 через 5 лет наблюдения; ФВ ЛЖ, оставаясь умеренно сниженной, составила 38–47% через 1 год и 43% через 5 лет; при этом z-score левого предсердия (ЛП) сохранялся выше нормальных значений с некоторым уменьшением с +2,27 до +2,02 через 1 год и увеличением до +2,2 к 5-му году наблюдения. При первичной госпитализации у 7 (8,8%) пациентов обращала на себя внимание выраженная митральная недостаточность, у 13 (16,9%) — умеренная.
У 7 детей наблюдалось сочетание КМП с врождёнными пороками сердца: по 2 случая открытого артериального протока, дефекта межжелудочковой перегородки и дефекта межпредсердной перегородки, 1 — частичный аномальный дренаж лёгочных вен. За время наблюдения 5 из них проведена хирургическая коррекция.
При проведении СМ-ЭКГ во время первичной госпитализации в наш Центр гемодинамически значимых нарушений ритма сердца не обнаружено. Тем не менее у части пациентов выявлены различные нарушения сердечного ритма: атриовентрикулярная блокада 1 степени диагностирована у 7 (8,8%) детей, признаки предвозбуждения желудочков — у 17 (21,2%), эпизоды суправентрикулярной тахикардии — у 3 (3,8%), а желудочковая тахикардия без нарушения самочувствия — у 1 (1,2%).
По результатам молекулярно-генетического исследования пациентов с MYH7-обусловленной КМП чаще всего встречались мутации в экзонах 7, 22 и 23 (n = 10, 11 и 8 соответственно), несколько реже — в экзонах 8, 21 и 37 (n = 5 в каждом). Наиболее часто выявлялся вариант c.602T>C (экзон 7) — у 8 (9,6%) пациентов, их клинические характеристики уже были представлены нами ранее [24]. Вторым по частоте был вариант c.2678C>T (экзон 22), идентифицированный у 5 (6%) детей, трижды встречались варианты c.1106G>A (экзон 12), c.2330G>A и c.2710C>T (оба в экзоне 21), c.709C>T (экзон 8), дважды — c.2660T>G и c.2711G>A в экзонах 22 и 23 соответственно. Анализ клинического течения заболевания у пациентов с MYH7-обусловленной КМП выявил взаимосвязь между локализацией генетического варианта и тяжестью патологии. Так, наибольшая степень резистентности к консервативной терапии ХСН наблюдалась при нуклеотидных вариантах, расположенных в экзонах 5, 19–23.
За период наблюдения у 2 (2,5%) пациентов выявлены варианты c.445G>A в экзоне 5 и c.2647G>A в экзоне 22), зарегистрирован летальный исход (средний возраст — 11 мес). Значение как 5-, так и 10-летней выживаемости составило 97,4%. Ортотопическая трансплантация сердца была проведена 1 пациенту с вариантом c.602T>C, у остальных 7 пациентов с данным вариантом отмечено благоприятное течение заболевания. Хирургическая коррекция в связи с выраженной митральной недостаточностью проведена 5 пациентам: в 2 случаях — пластика (у пациентов с вариантами c.2330G>A в экзоне 21 и c.1106G>A в экзоне 12), в 3 — протезирование (у детей с вариантами c.2678C>T и c.2647G>A в экзоне 22, c.5385C>A в экзоне 37).
При анализе экстракардиальных проявлений особого внимания заслуживают неврологические нарушения. У 3 пациентов с MYH7-обусловленной КМП (с.1180G>A, c.5655+2T>C, c.3359_3370dup) были диагностированы миопатии. Примечательно, что проведённые молекулярно-генетические исследования не выявили у этих детей других вариантов, которые могли бы объяснить развитие неврологической симптоматики.
Приведём 3 клинических примера пациентов с сочетанием MYH7-обусловленной ДКМП и миопатией.
Описание клинического случая № 1
Девочка А., 2022 г.р., с внутриутробно выявленной ДКМП, обусловленной вариантом с.1180G>A в экзоне 13 гена MYH7 (chr14:23898515). Ребёнок от считающих себя здоровыми, отказавшихся от генетического обследования родителей, от 4-й беременности (в анамнезе — 1 срочные роды и 2 прерывания беременности) когда на 28-й неделе были выявлены дилатация полостей сердца, регургитация на атриовентрикулярных клапанах, от 2-х родов на 33-й неделе. При рождении масса 2900 г, оценка по Апгар 4/6/6 баллов, состояние тяжёлое, обусловленное сердечной недостаточностью и недоношенностью. С раннего возраста обращала на себя внимание задержка моторного и психо-предречевого развития: с 3,5 мес удерживает голову, с 9 мес — переворачивается, к 12 мес — гулит.
По данным ЭхоКГ, проведённой после рождения, выявлена дилатация полости ЛЖ с КДР 23 мм с последующим увеличением до 40 мм к 6 мес, дилатация ЛП, снижение ФВ до 19%. По СМ-ЭКГ частая суправентрикулярная экстрасистолия до 14 тыс в сутки. Повышение уровня NTproBNP до 3823 пг/мл. Врождённые пороки сердца были исключены. Проводилась кардиотоническая и респираторная поддержка, инициирована терапия ХСН, на фоне которой параметры ЭхоКГ оставались прежними.
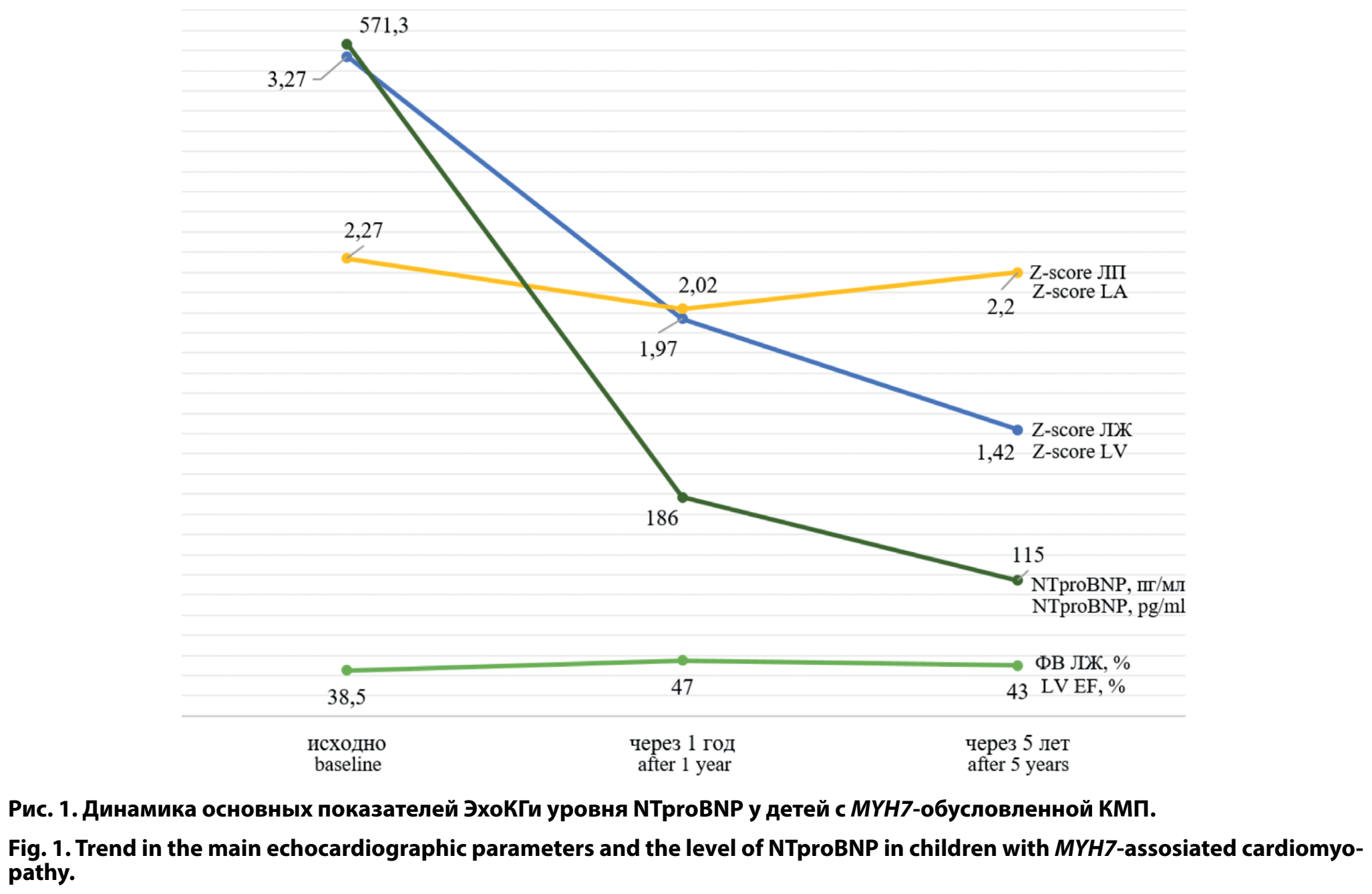
В 15 мес девочка впервые госпитализирована в кардиологическое отделение Центра по системе телемедицины. При поступлении обращало на себя внимание низкое, дисгармоничное физическое развитие (вес 8,3 кг, рост 74 см), диффузная мышечная гипотония. По результатам обследования сохранялся высокий уровень NTproBNP — 2889 пг/мл, отмечалось снижение параметра креатинфосфокиназы до 11 ЕД/л. По данным ЭхоКГ отмечена дилатация ЛЖ до 3,6 z-score и ЛП до 4,2 z-score, снижение ФВ до 13% по Симпсон, давление в системе лёгочной артерии составило 50 мм рт. ст., выявлены признаки некомпактного миокарда ЛЖ (рис. 2). По результатам рентгенографии органов грудной клетки кардиоторакальный индекс составил 71%. При проведении ЭКГ-исследования, включая СМ-ЭКГ, значимых нарушений ритма сердца и проводимости не регистрировалось. За период пребывания в отделении девочке была скорректирована медикаментозная терапия сердечной недостаточности. При контрольном плановом обследовании состояния через 6 мес получены следующие данные: уровень NTproBNP 2469 пг/мл; сохранялись эхо-признаки ремоделирования по дилатационному фенотипу без значимого изменения параметров; нормализовалось давление в системе лёгочной артерии; уменьшились рентгенологические размеры сердца (кардиоторакальный индекс 69%); по данным СМ-ЭКГ обращали на себя внимание эпизоды синусовой тахикардии без эктопической активности. Терапия была скорректирована в соответствии с результатами обследования.
В 21 мес девочка была консультирована неврологом. При осмотре обращала на себя внимание задержка психоречевого развития: экспрессивная речь — лепет, несколько простых слов-слогов, манипулятивная деятельность с игрушками. Задержка моторного развития: голову держит, поворачивается на бок, переворачивается со спины на живот, при тракции за руки группируется, посаженная сидит, самостоятельно не садится, при вертикализации есть опора на стопы, самостоятельно не встаёт, не ходит. В неврологическом статусе со стороны черепных нервов — без очаговой патологии. Отмечается диффузная мышечная гипотония, гипермобильный синдром в крупных и мелких суставах, сухожильные рефлексы и надкостничные рефелексы с верхних и нижних конечностей живые, D = S. Снижение объёма активных движений, преимущественно в верхнем плечевом поясе. Данные симптомы характерны для миопатических проявлений.
Игольчатая электромиография в настоящее время не проведена, продолжено наблюдение за ребёнком.
Описание клинического случая № 2
Девочка Б., 2022 г.р., с MYH7-обусловленной ДКМП, при генетическом обследовании которой в интроне 38 был выявлен de novo вариант c.5655+2T>C (chr14:23414005A>G). Ребенок от 3-й беременности (в анамнезе — антенатальная гибель плода), 2-х самостоятельных срочных родов, вес при рождении 3650 г, длина тела 54 см, оценка по Апгар 8/9 баллов. Раннее психомоторное развитие с задержкой: в 6 мес — голову не держит, не садится, при тракции за руки — не группируется, начала переворачиваться на бок, опоры на стопы нет. В 11 мес начала самостоятельно переворачиваться на живот, при осмотре в 14 мес самостоятельно не садится, сидит посаженная, в речевом развитии — слоги, «ма-ма».
В неврологическом статусе со стороны черепных нервов очаговой патологии не выявлено. Диффузная мышечная гипотония с сохранными симметричными сухожильными и надкостничными рефлексами с верхних и нижних конечностей, ограничение объёма активных движений. Таким образом, у пациентки имеется миопатический симптомокомплекс. В 11 мес по месту жительства проведена игольчатая электромиография, выявлены признаки первично-мышечных изменений мышц верхних и нижних конечностей со снижением амплитуды и длительности потенциалов двигательных единиц (ПДЕ) исследуемых мышц (слева длительность ПДЕ 5,21 мс, амплитуда 137 мкВ, справа — 4,12 мс и 153 мкВ) и изменением паттерна активации по миопатическому типу.
Наблюдается ортопедом по поводу вальгусной установки стоп, сколиотического типа нарушения осанки.
Симптомы сердечной недостаточности впервые выявлены на 3-й неделе жизни, по данным ЭхоКГ — дилатация ЛЖ со снижением ФВ до 20%, повышение уровня NTproBNP до 4397 пг/мл. Была начата терапия сердечной недостаточности, и впервые в 3 мес девочка направлена в наш Центр по системе телемедицины. По результатам обследования оставался повышенным уровень NTproBNP до 1760 пг/мл. По данным ЭхоКГ сохранялись дилатация ЛЖ до 4 Z-score и ЛП до 2 Z-score, повышенная трабекулярность апикальных сегментов ЛЖ, снижение ФВ до 30% по Симпсон. При проведении СМ-ЭКГ выявлена δ-волна в грудных отведениях. Проведена коррекция терапии ХСН, на фоне которой к 1 году 10 мес отмечено снижение маркера перегрузки миокарда до 124 пг/мл; по ЭхоКГ нормализовались размеры левых камер сердца (Z-score ЛЖ и ЛП 1,3), улучшилась сократительная способность с ФВ 52%. Продолжено наблюдение за ребёнком.
Описание клинического случая № 3
Мальчик В., 2013 г.р., у которого на 1-е сутки жизни выявлен некомпактный миокард и ДКМП. Генетическое обследование выявило в экзоне 27 гена MYH7 de novo вариант c.3359_3370dup (chr14:23420214-23420225dup).
Мальчик от 2-й беременности, протекавшей с угрозой прерывания на 11–12-й неделе, 2-х родов на 39-й неделе. Масса при рождении 4150 г, длина тела 60 см, оценка по Апгар 7/7 баллов. Раннее моторное развитие с незначительной темповой задержкой. С 4 лет наблюдался ортопедом по поводу нарушения осанки.
По результатам представленных ЭхоКГ в 9 и 10 лет сохранялся некомпактный миокард без указания соотношения, дилатация ЛЖ и обоих предсердий, снижение ФВ до 45%. По представленным данным, мальчик получал медикаментозную терапию в соответствии с клиническими рекомендациями по лечению ХСН у детей.
Регулярно наблюдался неврологом, проводилась верификация диагноза, за время наблюдения отмечалось однократное повышение уровня КФК до 2 норм. Игольчатая электромиография в 10 лет показала признаки умеренной дисфункции nn peroneus и незначительной дисфункции nn tibialis по типу аксонопатии (моторные волокна), сенсорная проводимость не нарушена.
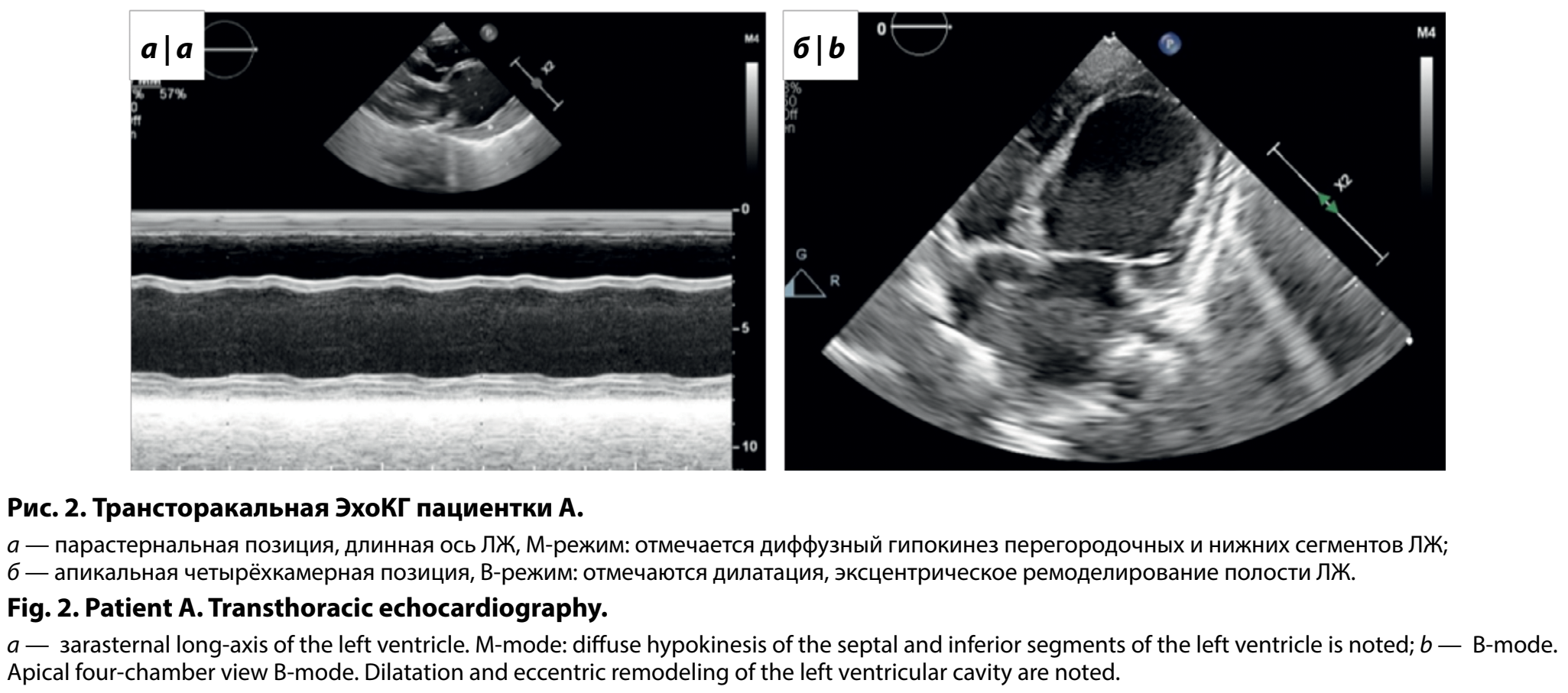
В 11 лет впервые направлен в Центр, проведено МРТ мышц (рис. 3) — картина билатеральной атрофии мышц бедра с неравномерной жировой инволюцией, более выражены изменения задней группы мышц, минимально визуализируется билатеральная атрофия мышц с неравномерной жировой инволюцией в передней группе мышц голени (затронуты большеберцовая и малоберцовая мышцы и сгибатели пальцев стопы). Неврологический осмотр: черепные нервы без очаговой патологии. В двигательной сфере: объём пассивных и активных движений не ограничен. Мышечный тонус снижен в руках и ногах. Мышечная сила в верхних конечностях снижена, достоверно до 3–4 баллов, в нижних конечностях — до 3 баллов в дистальных отделах, до 4 баллов в проксимальных отделах. Не может ходить на пятках. Сухожильные рефлексы с рук равномерно снижены, коленные — с двух сторон отсутствуют, ахилловы — вызываются, D = S. Клонусов стоп нет. Патологические стопные симптомы отрицательные с 2 сторон. Брюшные рефлексы живые. Использует приёмы Говерса при вертикализации (не может самостоятельно подняться с пола из положения лежа/сидя). Поднимается по лестнице, опираясь на поручень. Походка — степпаж. Грубых координаторных нарушений нет. Пальце-носовую пробу выполняет с лёгкой дисметрией. Нарушений чувствительности нет. Тазовые функции не нарушены.
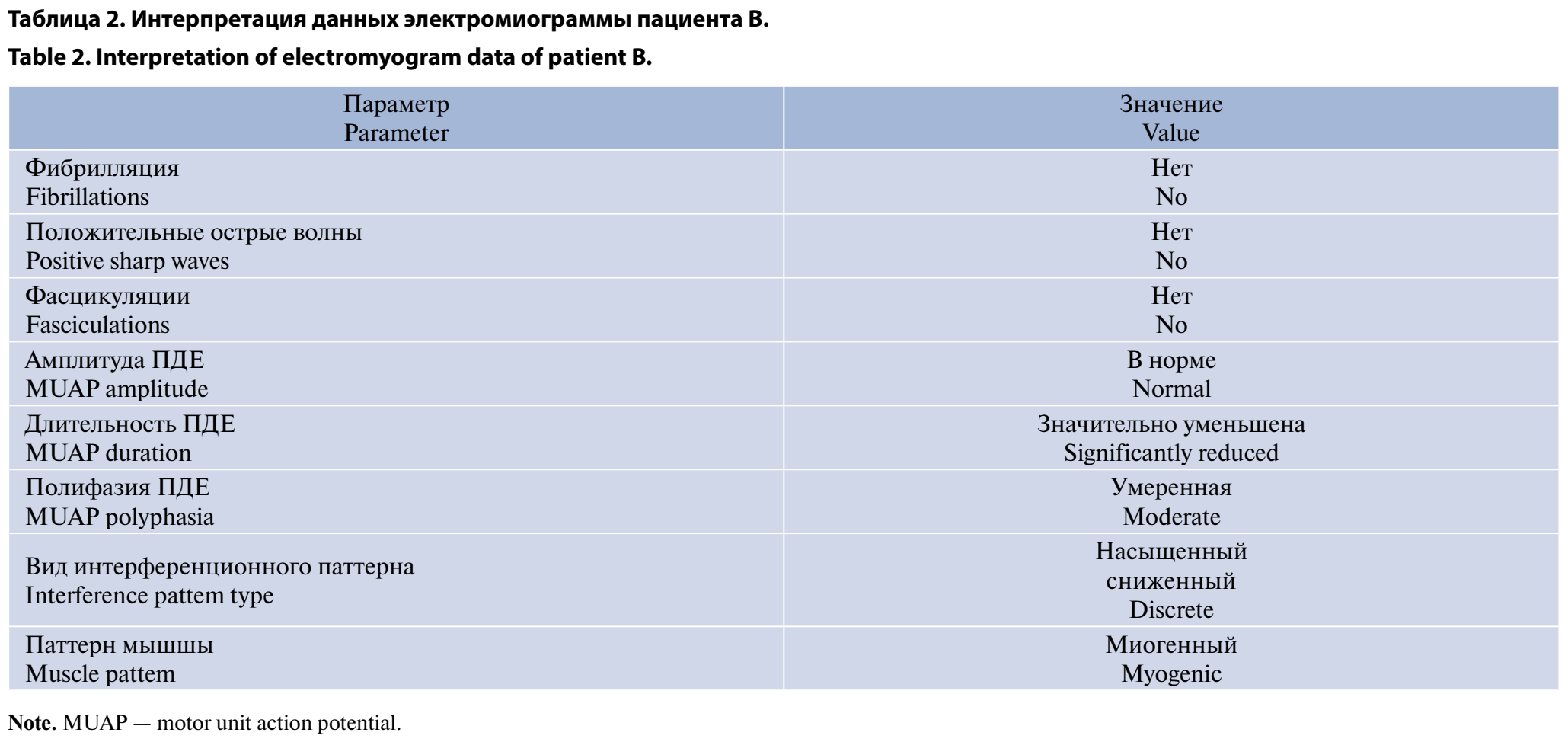
В нашем Центре проведена игольчатая электромиография (рис. 4): при игольчатом исследовании латеральной порции четырёхглавой мышцы бедра слева зарегистрированы ПДЕ со средней длительностью 7,1 мс (норма 9,3 мс), т. е. снижение на 23,7%. Доминируют ПДЕ с достоверно укороченной длительностью. Средняя амплитуда — 489 мкВ, среднее число фаз 4, полифазных ПДЕ — 15%. В покое спонтанная активность не выявлена. При проведении турн-амплитудного анализа больше половины (60%) полученных значений расположены вне нормативной облачной диаграммы. Паттерн рекрутирования ПДЕ в каждой исследованной мышце — полный (табл. 2). Выявленные признаки указывают на мышечный тип поражения (средняя длительность ПДЕ значимо снижена, при турн-амплитудном анализе больше половины значений расположены вне нормативной облачной диаграммы, во всех тестированных мышцах паттерн рекрутирования ПДЕ полный). При этом скорость распространения возбуждения по периферическим нервам ног не снижена.
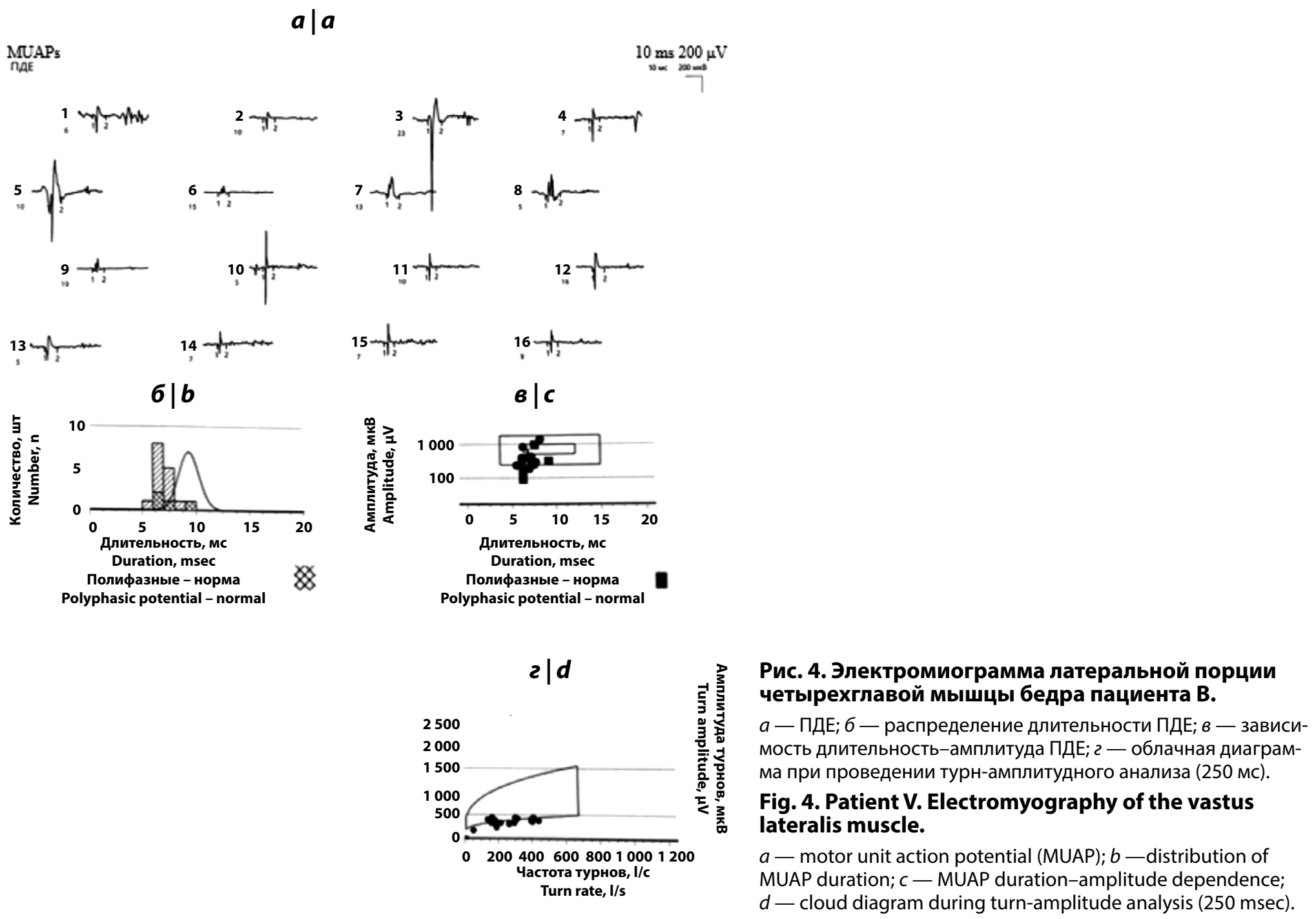
Ребёнок консультирован генетиком: по результатам клинико-анамнестических данных выявлены MYH7-обусловленная миопатия, аутосомно-доминантная, в частности, дистальная миопатия Лэинга (OMIM #160500).
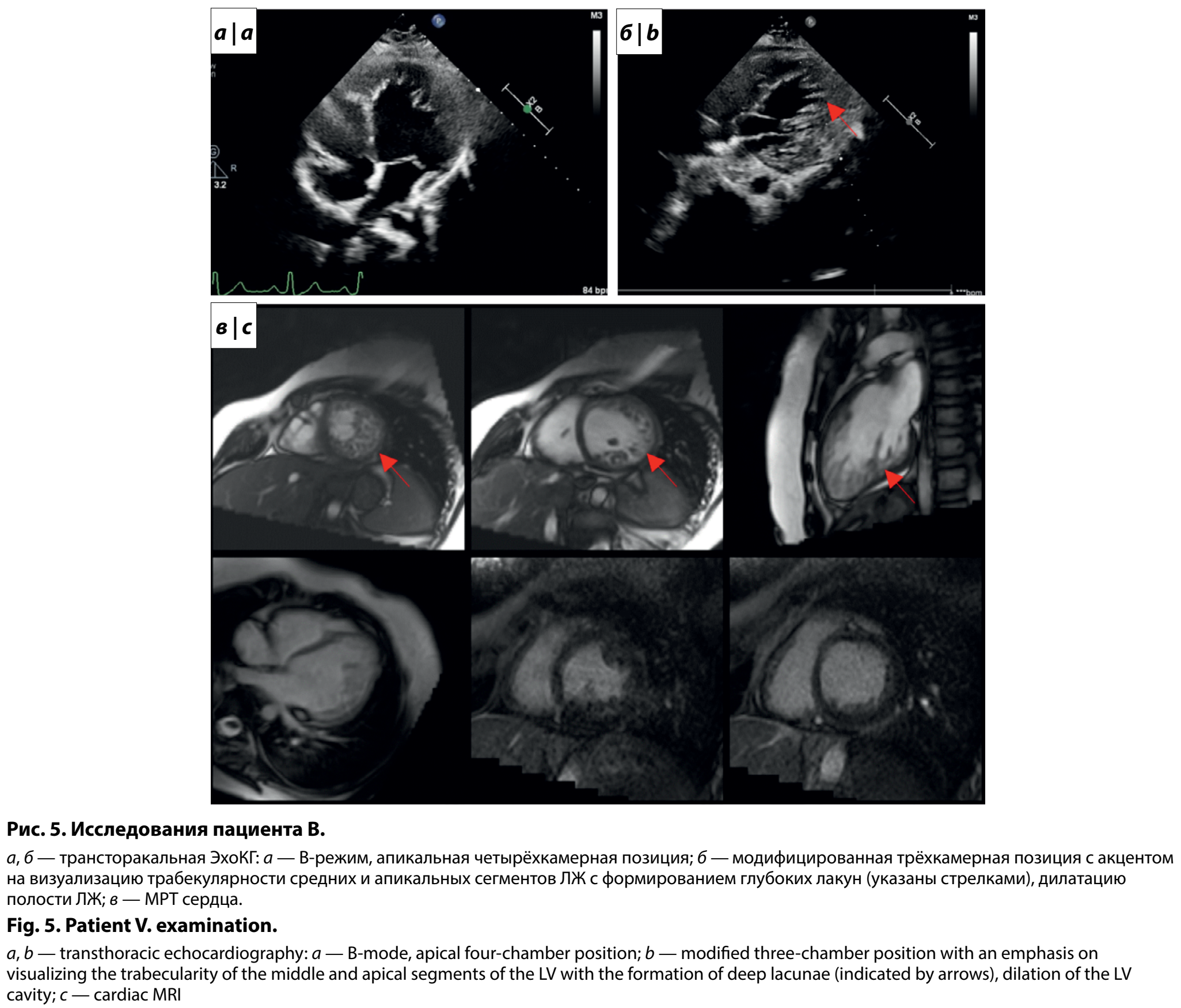
По результатам обследования в кардиологическом отделении Центра в 11 лет отмечен нормальный уровень NTproBNP (12,7 пг/мл). По ЭхоКГ выявлены признаки некомпактного миокарда апикальных и средних сегментов ЛЖ в соотношении 22:6 мм, сферичность полости ЛЖ (индекс сферичности 0,9), пограничный размер полости ЛЖ (Z-score = 2), снижение ФВ по Симпсон до 43%, диастолическая дисфункция по 1-му типу (рис. 5, а, б). По результатам СМ-ЭКГ отмечена склонность к тахикардии, δ-волна в большинстве отведений, значимых нарушений ритма сердца и пауз ритма не регистрировалось. Проведено МРТ сердца (рис. 5, в), подтвердившее некомпактный миокард, снижение функциональных параметров обоих желудочков (ФВ ЛЖ = 39%, правого желудочка — 51%), гипокинез перегородки на всем протяжении, зон фиброза не определялось. Подростку подобрана терапия сердечной недостаточности, продолжено динамическое наблюдение за мальчиком.
Обсуждение
Как один из преобладающих вариантов ремоделирования дилатационный фенотип ассоциирован с высокой заболеваемостью и смертностью, что требует его тщательного изучения [3, 25, 26]. Согласно последним исследованиям, наследственные факторы ответственны более чем за 30% случаев заболевания, при этом основную роль играют патогенные варианты генов саркомерных белков [2, 3, 27].
Ген MYH7 располагается на 14-й хромосоме, содержит 40 экзонов и кодирует β-тяжёлую цепь миозина, обеспечивающую сердечные сокращения. Его мутации нарушают работу саркомера, приводя к миокардиальной дисфункции [27–29]. Исследования среди взрослых больных показывают, что MYH7 лидирует среди генов, вызывающих КМП (около 35% случаев), занимая центральное место в диагностических алгоритмах [4, 29, 30]. Другие работы, проведённые во взрослой популяции, демонстрируют меньшую представленность вариантов в гене MYH7 (менее 10%), называя более распространёнными варианты в генах TTN (до 20% случаев) и LMNA (10–15%) [3, 8, 31–33]. Однако полученные нами данные выявили существенное отличие в генетическом ландшафте у детей — более 40% случаев были обусловлены патогенными вариантами гена MYH7, что превышает показатели, описанные в литературе. Более чем у 80% наших пациентов заболевание выявлено на 1-м году жизни. Полученные данные могут говорить о более раннем дебюте MYH7-обусловленных КМП, что подчёркивает значимость ранней диагностики КМП и необходимость тщательного врачебного контроля за детьми раннего возраста.
Частота семейных форм MYH7-обусловленной ДКМП у детей составляет 38%, что согласуется с современными представлениями о ключевой роли наследственности в развитии заболевания. Этот показатель в детской популяции выше, чем у взрослых (20–25%) [26], что, вероятно, свидетельствует о большей генетической обусловленности у детей. Важно отметить, что во многих семьях наблюдались неполная пенетрантность и вариабельная экспрессивность, что затрудняет диагностику и требует расширенного скрининга даже у бессимптомных родственников. Эти выводы согласуются с рекомендациями Европейского общества кардиологов по ведению наследственных КМП [3].
ДКМП традиционно характеризуется прогрессирующим течением с развитием ХСН, частыми аритмиями и высоким риском неблагоприятных исходов [3, 7, 31]. Однако в нашем исследовании пациенты с патогенными вариантами в гене MYH7 на фоне адекватной медикаментозной терапии ХСН продемонстрировали относительно благоприятную динамику: нормализовался размер ЛЖ, улучшилась сократительная способность, снизился уровень NTproBNP, не регистрировалось клинически значимых нарушений сердечного ритма, а также высокую 5- и 10-летнюю выживаемость. Наши данные согласуются с результатами исследования 147 взрослых пациентов, где было показано, что у 18% пациентов заболевание впервые проявилось до 18 лет, при этом течение болезни характеризовалось относительно благоприятным прогнозом, частота жизнеугрожающих аритмий была низкой (до 2%), а у многих пациентов наблюдалось обратное развитие ремоделирования миокарда [34]. Эти данные позволяют предположить существование более доброкачественного фенотипа заболевания у пациентов с вариантами в гене MYH7. На основании регистрируемого в течение 5 лет увеличения дилатации ЛП можно предположить, что некомпактный миокард выступает в роли фактора, провоцирующего нарушения диастолической функции в отдалённом периоде.
R.H. Pignatelli и соавт. сообщают, что у 25–40% детей с MYH7-обусловленной ДКМП обнаруживается некомпактный миокард [35]. Наше исследование показало более чем 85-процентное сочетание дилатационного фенотипа с признаками некомпактности.
Согласно данным литературы, патогенные варианты гена MYH7 могут приводить как к изолированным миопатиям, так и к их сочетанию с различными фенотипами КМП, при этом большинство миопатических форм ассоциированы с мутациями в экзонах 35–38 [14–18]. В нашей работе выявлены 3 случая КМП с неоднородной неврологической симптоматикой, что, вероятно, связано с разной локализацией мутаций в гене (экзоны 13, 27 и интрон 38) и их типом (миссенс, дупликация и сплайсинг). У пациента с вариантом в экзоне 27 течение КМП было более мягким, тогда как у двух других детей с мутациями в экзоне 13 и интроне 38 преобладали проявления сердечной недостаточности. Подобные случаи требуют мультидисциплинарного подхода к диагностике и лечению заболевания у детей.
Заключение
Исследование подтверждает важную роль вариантов гена MYH7 в развитии ДКМП у детей. MYH7-обусловленные формы заболевания характеризуются ранним дебютом, частым сочетанием с некомпактным миокардом, относительно благоприятным прогнозом при медикаментозном лечении. Однако пациенты с мутациями в экзонах 19–23 чаще нуждаются в хирургической коррекции митральной недостаточности. Своевременная молекулярно-генетическая диагностика позволяет не только верифицировать диагноз, но и проводить ранний скрининг среди родственников, прогнозировать течение болезни и оптимизировать тактику ведения пациентов. Важным аспектом является систематизация клинико-генетических характеристик, которая создаёт основу для разработки персонализированных подходов к терапии. Наличие экстракардиальных проявлений при КМП диктует необходимость комплексного междисциплинарного наблюдения за пациентами.
1. Леонтьева И.В. Проблемы современной диагностики и лечения дилатационной кардиомиопатии у детей. Российский вестник перинатолологии и педиатрии. 2018; 63(2): 7–15. https://doi.org/10.21508/1027-4065-2018-63-2-7-15 https://elibrary.ru/lbmyyh
2. Pugh T.J., Kelly M.A., Gowrisankar S., Hynes E., Seidman M.A., Baxter S.M., et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014; 16(8): 601–8. https://doi.org/10.1038/gim.2013.204
3. Arbelo E., Protonotarios A., Gimeno J.R., Arbustini E., Barriales-Villa R., Basso C., et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023; 44(37): 3503–626. https://doi.org/10.1093/eurheartj/ehad194
4. Савостьянов К.В., Намазова-Баранова Л.С., Басаргина Е.Н., Вашакмадзе Н.Д., Журкова Н.В., Пушков А.А. и др. Новые варианты генома российских детей с генетически обусловленными кардиомиопатиями, выявленные методом массового параллельного секвенирования. Вестник Российской академии медицинских наук. 2017; 72(4): 242–53. https://doi.org/10.15690/vramn872 https://elibrary.ru/zfourx
5. Towbin J.A., Lowe A.M., Colan S.D., Sleeper L.A., Orav E.J., Clunie S., et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006; 296(15): 1867–76. https://doi.org/10.1001/jama.296.15.1867
6. Tsatsopoulou A., Protonotarios I., Xylouri Z., Krommydas A., Gatzoulis K.A., Tsioufis C., et al. Cardiomyopathies in children: An overview. Hellenic J. Cardiol. 2023; 72: 43–56. https://doi.org/10.1016/j.hjc.2023.02.007
7. Weintraub R.G., Semsarian C., Macdonald P. Dilated cardiomyopathy. Lancet. 2017; 390(10092): 400–14. https://doi.org/10.1016/S0140-6736(16)31713-5
8. McNally E.M., Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 2017; 121(7): 731–48. https://doi.org/10.1161/CIRCRESAHA.116.309396
9. Stroeks S.L.V.M., Lunde I.G., Hellebrekers D.M.E.I., Claes G.R.F., Krapels I.P., Vanhoutte E.K., et al. Prevalence and clinical consequences of multiple pathogenic variants in dilated cardiomyopathy. Circ. Genom. Precis. Med. 2023; 16(2): e003788. https://doi.org/10.1161/CIRCGEN.122.003788
10. Савостьянов К.В. Современные алгоритмы генетической диагностики редких наследственных болезней у российских пациентов. М.; 2022. https://elibrary.ru/rduzgh
11. Eldemire R., Mestroni L., Taylor M.R.G. Genetics of dilated cardiomyopathy. Annu. Rev. Med. 2024; 75: 417–26. https://doi.org/10.1146/annurev-med-052422-020535
12. Chen S.N., Mestroni L., Taylor M.R.G. Genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2021; 36(3): 288–94. https://doi.org/10.1097/HCO.0000000000000845
13. Ware S.M., Wilkinson J.D., Tariq M., Schubert J.A., Kim C.E., DePalma S.R., et al. Genetic causes of cardiomyopathy in children: first results from the pediatric cardiomyopathy genes study. J. Am. Heart Assoc. 2021; 10(9): e017731. https://doi.org/10.1161/JAHA.120.017731
14. Tajsharghi H., Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013; 125(1): 3–18. https://doi.org/10.1007/s00401-012-1024-2
15. Fiorillo C., Astrea G., Savarese M., Cassandrini D., Brisca G., Trucco F., et al. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J. Rare Dis. 2016; 11(1): 91. https://doi.org/10.1186/s13023-016-0476-1
16. Naderi N., Mohsen-Pour N., Nilipour Y., Pourirahim M., Maleki M., Kalayinia S. A novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy. BMC Cardiovasc. Disord. 2023; 23(1): 487. https://doi.org/10.1186/s12872-023-03538-8
17. Naddaf E., Waclawik A.J. Two families with MYH7 distal myopathy associated with cardiomyopathy and core formations. J. Clin. Neuromuscul. Dis. 2015; 16(3): 164–9. https://doi.org/10.1097/CND.0000000000000069
18. Bahout M., Severa G., Kamoun E., Bouhour F., Pegat A., Toutain A., et al. MYH7-related myopathies: clinical, myopathological and genotypic spectrum in a multicentre French cohort. J. Neurol. Neurosurg. Psychiatry. 2025; 96(5): 453–61. https://doi.org/10.1136/jnnp-2024-334263
19. Вайханская Т.Г., Сивицкая Л.Н., Курушко Т.В., Даниленко Н.Г., Сташук Г.А., Пилецкий К.В. и др. Дилатационная кардиомиопатия: новый взгляд на проблему. Российский кардиологический журнал. 2019; 24(4): 35–47. https://doi.org/10.15829/1560-4071-2019-4-35-47 https://elibrary.ru/tqsqaj
20. Кучер А.Н., Слепцов А.А., Назаренко М.С. Генетический ландшафт дилатационной кардиомиопатии. Генетика. 2022; 58(4): 371–87. https://doi.org/10.31857/S0016675822030080 https://elibrary.ru/lavjrv
21. Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б., Коновалов Ф.А., Масленников А.Б., Степанов В.А. и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2). Медицинская генетика. 2019; 18(2): 3–23. https://doi.org/10.25557/2073-7998.2019.02.3-23 https://elibrary.ru/jzljue
22. Human Gene Mutation Database (HGMD). Professional 2024.1. Available at: http://www.hgmd.cf.ac.uk
23. Burykina Yu., Chudakova D., Zharova O., Savostyanov K., Pushkov A., Polyakov A., et al. A unique case of a child with two rare hereditary diseases: familial dilated cardiomyopathy and arterial calcification. Int. J. Mol. Sci. 2025; 26(12): 5900. https://doi.org/10.3390/ijms26125900
24. Бурыкина Ю.С., Жарова О.П., Сильнова И.В., Басаргина Е.Н., Давыдова Ю.И., Пахомов А.В. и др. Описание кардиомиопатии с дилатационным фенотипом, обусловленной патогенным вариантом c.602T>C в гене MYH7, у восьми неродственных российских детей. Детские болезни сердца и сосудов. 2025; 22(3): 159–70. https://doi.org/10.24022/1810-0686-2025-22-3-159-170 https://elibrary.ru/djicrx
25. Verdonschot J.A.J., Hazebroek M.R., Krapels I.P.C., Barandiarán Aizpurua A., Willemsen R., Saris C.G.J., et al. Implications of genetic testing in dilated cardiomyopathy. Circ. Genom. Precis. Med. 2020; 13(5): 476–87. https://doi.org/10.1161/CIRCGEN.120.003031
26. Hershberger R.E., Hedges D.J., Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013; 10(9): 531–47. https://doi.org/10.1038/nrcardio.2013.105
27. Kamisago M., Sharma S.D., DePalma S.R., Solomon S., Sharma P., McDonough B., et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000; 343(23): 1688–96. https://doi.org/10.1056/NEJM200012073432304
28. Villard E., Duboscq-Bidot L., Charron P., Benaiche A., Conraads V., Sylvius N., et al. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur. Heart J. 2005; 26(8): 794–803. https://doi.org/10.1093/eurheartj/ehi193
29. Jefferies J.L., Towbin J.A. Dilated cardiomyopathy. Lancet. 2010; 375(9716): 752–62. https://doi.org/10.1016/S0140-6736(09)62023-7
30. Kelly M.A., Caleshu C., Morales A., Buchan J., Wolf Z., Harrison S.M., et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018; 20(3): 351–9. https://doi.org/10.1038/gim.2017.218
31. Lamounier Júnior A., Ferrari F., Max R., Ritt L.E.F., Stein R. Importance of genetic testing in dilated cardiomyopathy: applications and challenges in clinical practice. Arq. Bras. Cardiol. 2019; 113(2): 274–81. https://doi.org/10.5935/abc.20190144
32. Hsu D.T., Canter C.E. Dilated cardiomyopathy and heart failure in children. Heart Fail. Clin. 2010; 6(4): 415–32. https://doi.org/10.1016/j.hfc.2010.05.003
33. Rosenbaum A.N., Agre K.E., Pereira N.L. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat. Rev. Cardiol. 2020; 17(5): 286–97. https://doi.org/10.1038/s41569-019-0284-0
34. de Frutos F., Ochoa J.P., Navarro-Peñalver M., Baas A., Bjerre J.V., Zorio E., et al. Natural history of MYH7-related dilated cardiomyopathy. J. Am. Coll. Cardiol. 2022; 80(15): 1447–61. https://doi.org/10.1016/j.jacc.2022.07.023
35. Pignatelli R.H., McMahon C.J., Dreyer W.J., Denfield S.W., Price J., Belmont J.W., et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003; 108(21): 2672–8. https://doi.org/10.1161/01.CIR.0000100664.10777.B8
Аспирант ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: julia1907925@yandex.ru
Канд. мед. наук, ст. науч. сотр., врач-детский кардиолог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: sdvigova-natalya@yandex.ru
Доктор мед. наук, профессор, гл. науч. сотр., врач-детский кардиолог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: basargina@nczd.ru
Канд. мед. наук, ст. науч. сотр., врач-невролог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: globa@nczd.ru
Доктор мед. наук, гл. науч. сотр., врач-невролог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: alkurenkov@gmail.com
Канд. мед. наук, ст. науч. сотр. лаб. медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: ilya_zhanin@outlook.com
Канд. мед. наук, ведущ. науч. сотр. лаб. медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: pushkovAA@nczd.ru
Доктор биол. наук, нач. Медико-генетического центра, зав. лаб. медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: 7443333@gmail.com
Бурыкина Ю.С., Сдвигова Н.А., Басаргина Е.Н., Глоба О.В., Куренков А.Л., Жанин И.С., Пушков А.А., Савостьянов К.В. Одноцентровое исследование клинико-генетических характеристик дилатационного фенотипа кардиомиопатии, обусловленной вариантами в гене MYH7, у 80 неродственных детей. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):195-208. https://doi.org/10.46563/2686-8997-2025-6-4-195-208. EDN: mhpsyv
Burykina Yu.S., Sdvigova N.A., Basargina E.N., Globa O.V., Kurenkov A.L., Zhanin I.S., Pushkov A.A., Savostyanov K.V. A single-center study of the clinical and genetic characteristics of the dilated cardiomyopathy caused by MYH7 gene variants in eighty unrelated children. L.O. Badalyan Neurological Journal. 2025;6(4):195-208. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-195-208. EDN: mhpsyv

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















