


Содержание
Перейти к:
 Людмила Михайловна Кузенкова,
Алексей Львович Куренков,
Евгения Владимировна Увакина,
Дарья Андреевна Фисенко,
Владислав Владимирович Черников,
Елена Леонидовна Семикина,
София Георгиевна Попович,
Бэлла Ибрагимовна Бурсагова,
Оксана Валерьевна Глоба,
Наталья Владимировна Андреенко,
Луизат Муслимовна Абдуллаева,
Юлия Александровна Курова,
Надежда Сергеевна Адалимова,
Дарья Сергеевна Николенко,
Анастасия Андреевна Лялина,
Ольга Викторовна Комарова
Людмила Михайловна Кузенкова,
Алексей Львович Куренков,
Евгения Владимировна Увакина,
Дарья Андреевна Фисенко,
Владислав Владимирович Черников,
Елена Леонидовна Семикина,
София Георгиевна Попович,
Бэлла Ибрагимовна Бурсагова,
Оксана Валерьевна Глоба,
Наталья Владимировна Андреенко,
Луизат Муслимовна Абдуллаева,
Юлия Александровна Курова,
Надежда Сергеевна Адалимова,
Дарья Сергеевна Николенко,
Анастасия Андреевна Лялина,
Ольга Викторовна Комарова https://doi.org/10.46563/2686-8997-2025-6-4-182-194
EDN: bletts
Перейти к:
Введение. Использование генной терапии у пациентов со спинальной мышечной атрофией (СМА) привело к значительному улучшению прогнозов двигательного развития. Включение СМА в расширенный неонатальный скрининг в России позволило не только максимально рано выявлять пациентов, но и для части из них стала доступна генная терапия на пресимптоматической стадии (ПС) заболевания.
Цель исследования — оценить в условиях реальной клинической практики эффективность генной терапии при 2-летнем катамнестическом наблюдении за пациентами со СМА, получивших лечение препаратом онасемноген абепарвовек на СМА-ПС.
Материалы и методы. В исследование были включены 35 ребёнка со СМА, с генетически верифицированным диагнозом СМА без развития симптомов заболевания. Диагноз СМА был установлен при проведении пилотного проекта расширенного неонатального скрининга или с 01.01.2023 в рамках расширенного неонатального скрининга. Диагноз был верифицирован при проведении молекулярно-генетического исследования — у всех детей была выявлена делеция 7-го и/или 8-го экзонов гена SMN1 в гомозиготном состоянии. Все пациенты получили генную терапию препаратом онасемноген абепарвовек, средний возраст на момент проведения терапии составил 2,00 ± 0,94 мес (95% ДИ 1,68–2,32), min — 1,00, max — 5,00. Проводилась комплексная оценка клинических (основные этапы моторного развития по критериям ВОЗ, оценка по шкалам HINE-2 и CHOP-INTEND), электронейромиографических (латентность, амплитуда и площадь негативного пика дистального М-ответа при электрической стимуляции локтевого нерва на запястье, скорость распространения возбуждения по двигательным волокнам локтевого нерва на предплечье) и биохимических (уровни лёгких и тяжёлых цепей нейрофиламентов (НФ) в сыворотке крови) показателей у пациентов со СМА-ПС до инициации генной терапии и через 6, 12, 18 и 24 мес после её проведения.
Результаты. Пациенты со СМА-ПС в нашем исследовании как до, так и после проведения генной терапии имели моторное развитие в соответствии с возрастными нормами и достоверно не отличались от детей без неврологической патологии. При сравнительном анализе электронейромиографических показателей детей со СМА и группы здоровых детей установлено, что у пациентов ср СМА-ПС на фоне генной терапии статистически значимых различий не получено. При проведении сравнительного анализа уровней лёгких и тяжёлых цепей НФ в сыворотке крови пациентов со СМА-ПС до проведения генной терапии и 76 неврологически здоровых детей, полученных в недавнем исследовании, показано, что медиана уровня лёгких цепей НФ при СМА была достоверно (p < 0,001) выше: 6,00 [Q1; Q3 — 6,00; 31,43] и 4,00 [2,50; 9,57] пг/мл соответственно. В то же время уровень тяжёлых цепей НФ при сравнении пациентов со СМА-ПС до проведения генной терапии со здоровыми сверстниками достоверно не отличался. Применение онасемногена абепарвовека у пациентов со СМА-ПС приводило к статистически значимому снижению уровня лёгких цепей НФ в сыворотке крови. Максимальное снижение показателей НФ отмечалось через 3–6 мес после проведения генной терапии.
Заключение. Проведение генной терапии с применением препарата онасемноген абепарвовек у пациентов со СМА-ПС позволило предупредить развитие симптомов заболевания и формировать основные этапы моторного развития в соответствии с критериями ВОЗ.
Соблюдение этических стандартов. На проведение данного исследования было получено разрешение локального этического комитета ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России (протокол заседания ЛЭК № 10 от 06.10.2022).
Участие авторов:
Кузенкова Л.М., Куренков А.Л., Увакина Е.В., Фисенко Д.А. — концепция и дизайн статьи, написание текста, редактирование;
Черников В.В. — статистическая обработка данных, редактирование.
Все соавторы — редактирование, утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов в связи с публикацией данной статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Поступила 06.11.2025
Принята к печати 01.12.2025
Опубликована 31.01.2026
Кузенкова Л.М., Куренков А.Л., Увакина Е.В., Фисенко Д.А., Черников В.В., Семикина Е.Л., Попович С.Г., Бурсагова Б.И., Глоба О.В., Андреенко Н.В., Абдуллаева Л.М., Курова Ю.А., Адалимова Н.С., Николенко Д.С., Лялина А.А., Комарова О.В. Результаты двухлетнего комплексного наблюдения за пациентами со спинальной мышечной атрофией, получившими генную терапию препаратом онасемноген абепарвовек на пресимптоматической стадии заболевания. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):182-194. https://doi.org/10.46563/2686-8997-2025-6-4-182-194. EDN: bletts
Kuzenkova L.M., Kurenkov A.L., Uvakina E.V., Fisenko D.A., Chernikov V.V., Semikina E.L., Popovich S.G., Bursagova B.I., Globa O.V., Andreenko N.V., Abdullaeva L.M., Kurova J.A., Adalimova N.S., Nikolenko D.S., Lyalina A.A., Komarova O.V. Results of a two-year comprehensive follow-up in patients with spinal muscular atrophy who received gene therapy with onasemnogen abeparvovec at the presymptomatic stage of the disease. L.O. Badalyan Neurological Journal. 2025;6(4):182-194. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-182-194. EDN: bletts
Введение
Спинальная мышечная атрофия (СМА) — это наследственное нервно-мышечное заболевание, которое характеризуется поражением двигательных нейронов, что приводит к прогрессирующей мышечной слабости, дыхательным нарушениям, атрофии скелетных и бульбарных мышц [1].
С появлением патогенетической терапии СМА прогнозы значительно улучшились [2, 3]. В 2019 г. Управление по контролю за продуктами и лекарствами США одобрило препарат онасемноген абепарвовек (ОА). Проведение векторной генной терапии (ГТ) на основе аденоассоциированного вируса 9 с помощью однократного внутривенного введения позволяет доставлять полностью функциональную копию комплементарной ДНК гена SMN1 человека в клетки-мишени [4, 5]. Этот терапевтический подход всё шире применяется для лечения пациентов со СМА во всём мире.
Включение в России СМА в расширенный неонатальный скрининг позволило выявлять пациентов максимально рано и для части из них стала доступна генная терапия на пресимптоматической стадии (ПС) заболевания.
Цель исследования — оценить в условиях реальной клинической практики эффективность ГТ при 2-летнем катамнестическом наблюдении за пациентами со СМА, получивших лечение препаратом ОА на ПС заболевания.
Материалы и методы
В исследование были включены 35 детей с генетически верифицированным диагнозом СМА без развития симптомов заболевания. То есть, на момент включения в исследование и проведения лечения с применением ГТ все пациенты находились на ПС болезни. На проведение данного исследования было получено разрешение локального этического комитета ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России.
Диагноз СМА был установлен при проведении пилотного проекта расширенного неонатального скрининга или с 01.01.2023 в рамках расширенного неонатального скрининга. Диагноз был верифицирован при проведении молекулярно-генетического исследования — у всех детей была выявлена делеция экзонов 7 и/или 8 гена SMN1 в гомозиготном состоянии. Все пациенты получили генную терапию препаратом ОА, средний возраст на момент проведения терапии составил 2,0 ± 0,94 мес (95% ДИ 1,68–2,32), min — 1,00, max — 5,00.
Критерии включения:
Критерии исключения:
До включения в исследование детей осматривал педиатр для исключения заболеваний желудочно-кишечного тракта, дыхательной, сердечно-сосудистой и мочеполовой систем, не связанных с течением основного заболевания.
Для оценки двигательного, психического, предречевого и речевого развития всех пациентов осматривал невролог по стандартной методике [6], анализировались основные этапы двигательного развития с применением рекомендаций ВОЗ по 6 главным нормативным показателям достижения этапов моторного развития детей (сидение без поддержки, стояние с поддержкой, ползание на четвереньках, ходьба с поддержкой, стояние без поддержки, самостоятельная ходьба), моторные функции с применением шкал HINE-2 (шкала для короткого обследования на основе балльной системы для оценки двигательных функций детей в возрасте 2–24 мес) и CHOP-INTEND (Тест детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорождённых).
Всем детям, включённым в исследование, проводили электронейромиографию (ЭНМГ) до старта терапии и в течение последующих 2 лет. ЭНМГ выполняли на 2-канальном электронейромиографе «Нейро-МВП-Микро» («Нейрософт») с использованием стандартных биполярных одноразовых накожных электродов. Проводили тестирование двигательных волокон локтевого нерва, при этом регистрирующий электрод располагался над hypothenar в области проекции мышцы, отводящей 5-й палец кисти, референтный электрод — на 2-й фаланге 5-го пальца. Электрическую стимуляцию проводили в области проекции локтевого нерва на 1 см проксимальнее запястья в нижней трети предплечья по его внутреннему краю (1 точка) и в области локтя на (2 точка). Это позволило определить основные параметры негативного пика дистального М-ответа (при стимуляции в первой точке на запястье) — латентность, амплитуду и площадь, а также рассчитать скорость распространения возбуждения (СРВ) по двигательным волокнам дистальной части локтевого нерва — на предплечье.
У всех детей со СМА уровни лёгких и тяжёлых цепей нейрофиламента (НФ) в сыворотке крови были исследованы методом иммуноферментного анализа с применением следующих наборов реактивов: для определения протеина НФ 68 кДа (лёгкие цепи НФ) — «ELISA Kit for Neurofilament, Light Polypeptide (NEFL)», «Cloud-Clone Corp»; протеина НФ 200 кДа (тяжёлые цепи НФ) — «ELISA Kit for Neurofilament, Heavy Polypeptide (NEFH)», «Cloud-Clone Corp») с использованием оборудования «Tecan AustriaSunrise» и «Tecan Austria HydroFlex». Забор крови для исследования осуществляли в строго ограниченном количестве из периферических вен после оценки соматического статуса ребёнка, а также с учётом массы тела пациента.
Комплексную оценку клинических, нейрофизиологических и биохимических показателей у пациентов со СМА осуществляли до инициации ГТ и через 6, 12, 18 и 24 мес после её проведения.
К моменту завершения набора материала в исследование (март 2025 г.) из 35 пациентов 30 детей находились под наблюдением в течение минимум 12 мес, 23 ребёнка — в течение 18 мес, 12 детей — в течение 24 мес после проведения ГТ ОА. Такое распределение детей по времени наблюдения в нашем исследовании связано с тем, что неонатальный скрининг на СМА стартовал с 1 января 2023 г. и дети со СМА-ПС включались по мере их выявления при молекулярно-генетической диагностике.
Статистический анализ проводили с использованием программы «StatTech v. 4.6.1» («Статтех»). Количественные показатели оценивали на предмет соответствия нормальному распределению с помощью критерия Шапиро–Уилка, описывали с помощью средних арифметических величин (M) и стандартных отклонений (SD), границ 95% доверительного интервала (ДИ), медианы (Me) и нижнего и верхнего квартилей [Q1; Q3], минимума (min) и максимума (max). Категориальные данные описывали с указанием абсолютных значений (n) и процентных долей (%).
В связи с тем, что значения большинства показателей не подчинялись нормальному распределению, для анализа данных применяли непараметрические методы статистического анализа. Сравнение процентных долей при анализе четырёхпольных таблиц сопряжённости выполняли с помощью точного критерия Фишера. Сравнение двух групп по количественному показателю осуществляли с помощью U-критерия Манна–Уитни, сравнение трех и более групп — с помощью критерия Краскела–Уоллиса, апостериорные сравнения — с помощью критерия Данна с поправкой Холма. При сравнении количественных показателей в двух связанных группах использовали критерий Вилкоксона. Для сравнения 3 и более связанных групп применяли однофакторный дисперсионный анализ с повторными измерениями. Статистическую значимость изменений показателя в динамике оценивали с помощью критерия F Фишера. Апостериорный анализ проводили с помощью парного t-критерия Стьюдента с поправкой Холма. Различия считали значимыми при p < 0,05.
Результаты
Распределение пациентов по полу составило: 20 (57,1%) мальчиков и 15 (42,9%) девочек. Гендерное соотношение не имело значимых различий (р = 0,527). У большинства пациентов — 31 (88,6%) было 3 копии гена SMN2, у 4 (11,4%) детей — 2 копии.
Все дети, включённые в исследование на момент рождения, имели гестационный возраст 37–40 нед беременности, не имели отягощённого перинатального анамнеза: оценка по шкале Aпгар при рождении — 8 баллов и выше; вес — 2400 г и более; дети не имели признаков асфиксии и гипоксии, видимых пороков развития; не требовали перевода в отделение реанимации и интенсивной терапии, экстренного или планового хирургического лечения в раннем возрасте.
Пациенты, тестированные в возрасте от 0 до 30 дней, при неврологическом осмотре не имели общемозговых или менингеальных симптомов, патологии черепных нервов, изменений мышечного тонуса, повышения или снижения/отсутствия сухожильных рефлексов. У каждого ребёнка при осмотре определяли рефлексы новорождённого. При бодрствовании в положении лёжа на спине у всех детей наблюдалась спонтанная хаотичная двигательная активность верхними и нижними конечностями.
При неврологическом осмотре пациентов в возрасте от 1 до 6 мес не отмечалось общемозговых или менингеальных симптомов, патологии черепных нервов, изменений мышечного тонуса, повышения или снижения/отсутствия сухожильных рефлексов. К возрасту 2 мес большинство рефлексов новорождённого было редуцировано, отмечалось формирование комплекса оживления на эмоциональное общение со взрослым и элементы гуления.
Все пациенты, получившие генную терапию на ПС болезни, при катамнестическом наблюдении демонстрировали достижение всех основных двигательных навыков в соответствии с критериями ВОЗ, причём абсолютное большинство пациентов сформировали их в нормативные временные сроки. Только 1 (3,3%) ребёнок стал сидеть самостоятельно без поддержки и стоять самостоятельно позднее возрастных критериев — в 10 и 18 мес соответственно, а ещё 2 (6,6%) ребёнка стали «ходить самостоятельно» позднее возрастных критериев — в 20 и 21 мес соответственно.
При оценке пациентов перед стартом ГТ медиана значений по шкале HINE-2 составила 4,00 балла (2,00–4,50), min — 1,00, max — 19,00. Все дети, которые в процессе наблюдения достигли возраста самостоятельного хождения в соответствии с критериями ВОЗ (23 ребёнка), имели максимальное значение в 26 баллов по шкале HINE-2 к возрасту 18 мес (рис. 1). Медиана значений по шкале CHOP-INTEND перед стартом терапии ОА составила 57 баллов [54,00; 61,00], min — 47,00, max — 64,00. Через 6 мес после ГТ все пациенты достигли максимального значения 64 балла (рис. 1).
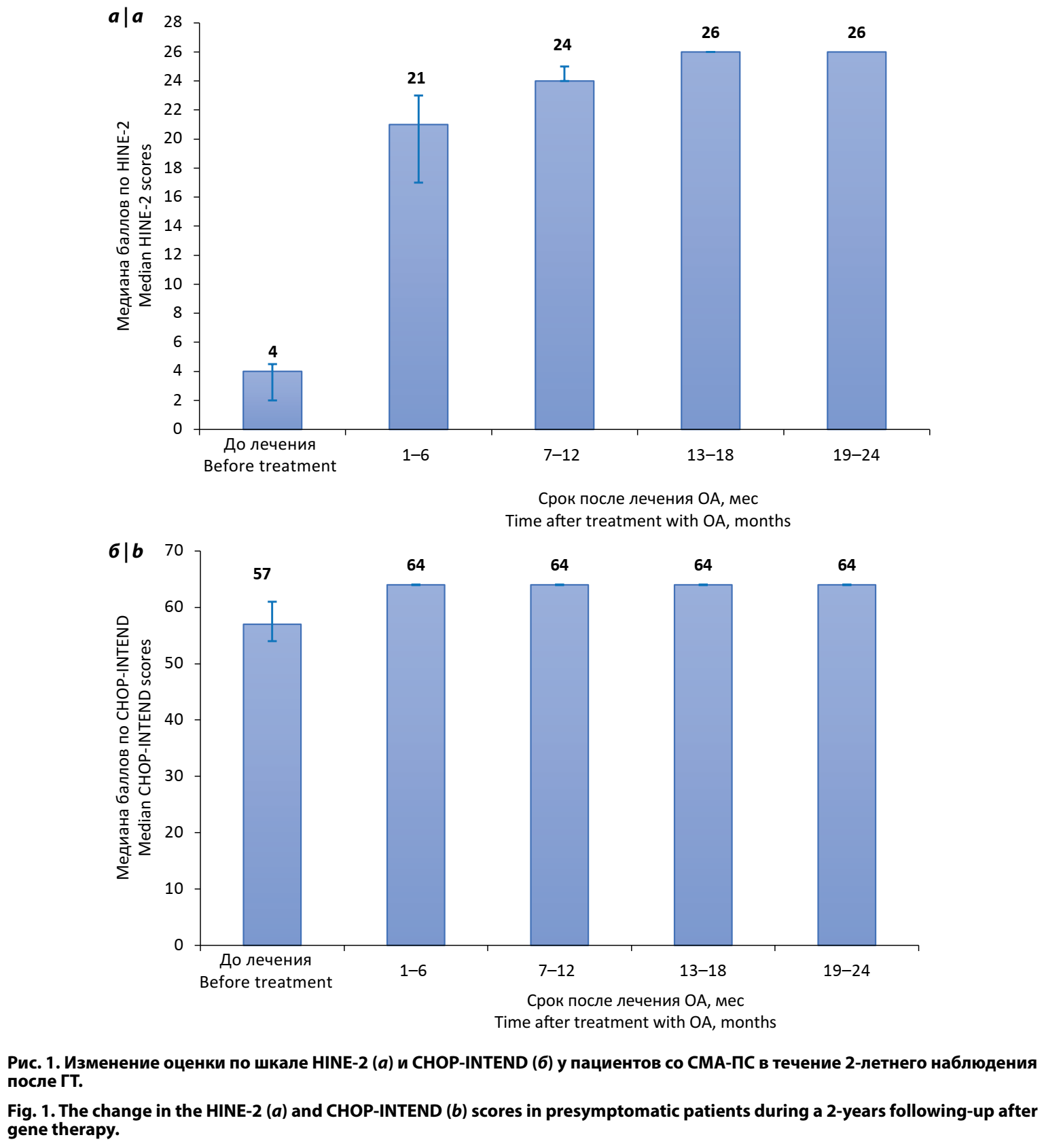
Таким образом, пациенты со СМА-ПС как до, так и после ГТ не имели нарушений моторного развития и сформировали двигательные навыки в соответствии с возрастными нормами, не отличаясь от детей без неврологической патологии. Применение ГТ на ПС заболевания позволяет у большинства пациентов предупредить развитие симптомов СМА.
Амплитуда, площадь и латентность негативного пика М-ответа мышцы, отводящей 5-й палец кисти при электрической стимуляции локтевого нерва в области запястья и СРВ по двигательным волокнам локтевого нерва на предплечье у пациентов со СМА-ПС представлены на рис. 2. У пациентов через 13–24 мес после ГТ было отмечено повышение основных показателей ЭНМГ: медианы амплитуда М-ответа — с 5,0 [4,3; 5,3] до 6,25 [5,20; 6,85] мВ; медианы площади М-ответа — с 8,90 [7,8; 10,1] до 11,60 [10,68; 13,22] мс × мВ; медианы СРВ — с 33,6 [32,4; 38,1] до 55,2 [52,8; 57,6] м/c.
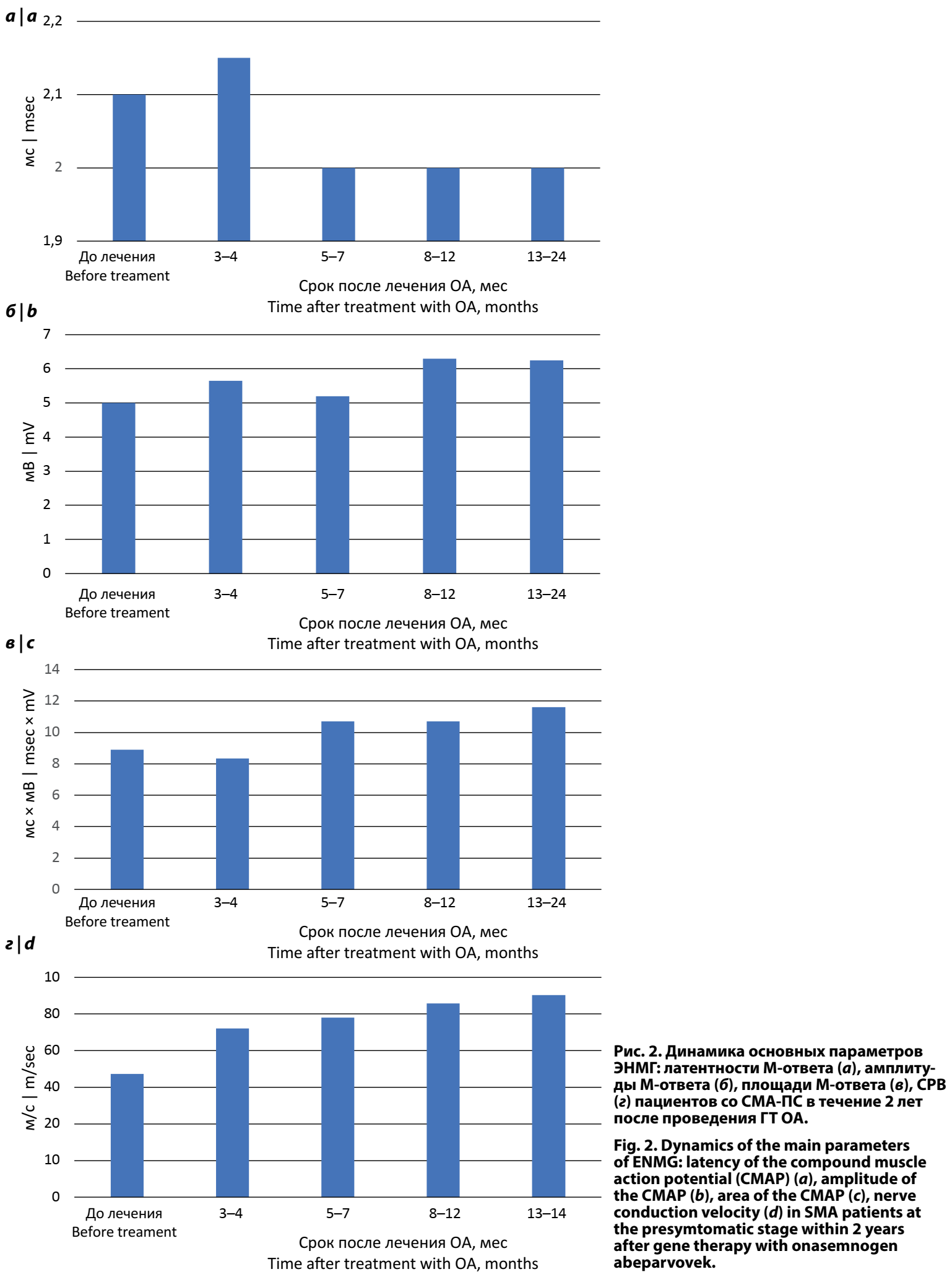
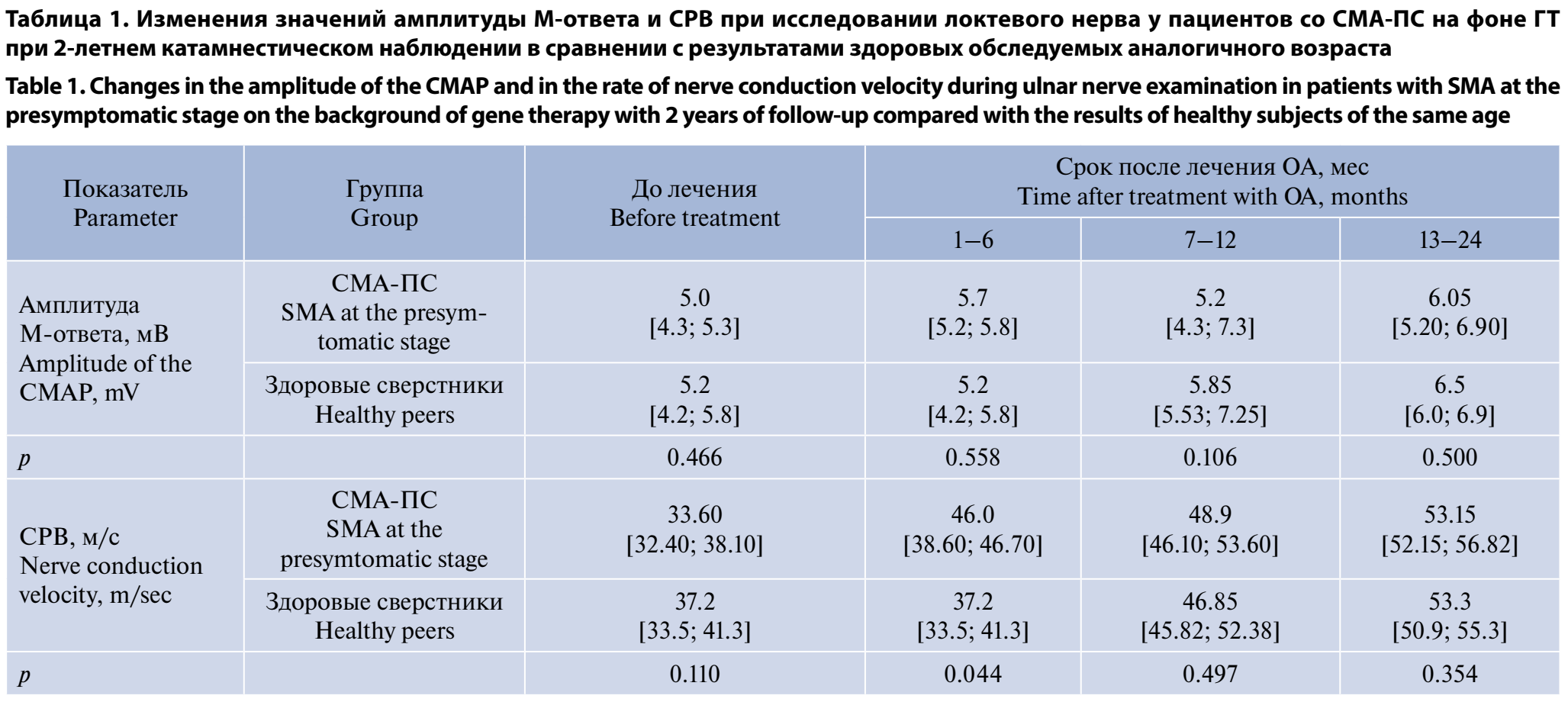
При сравнении основных параметров ЭНМГ (амплитуды М-ответа и СРВ по локтевому нерву) у пациентов со СМА-ПС с данными возрастной нормы, полученными при исследовании здоровых детей [7], достоверных отличий не было получено ни до старта терапии, ни в течение 2-летнего катамнестического наблюдения (табл. 1).
Таким образом, применение ГТ на ПС заболевания позволяет у большинства пациентов сохранять значения ЭНМГ-параметров в рамках возрастной нормы.
Полученные данные уровней лёгких и тяжёлых цепей НФ в сыворотке крови у детей со СМА-ПС до и через 3–6, 7–12 и 13–24 мес после ГТ не подчинялись нормальному распределению и описывались при помощи значений медианы, процентилей, минимума и максимума. Результаты представлены в табл. 2.
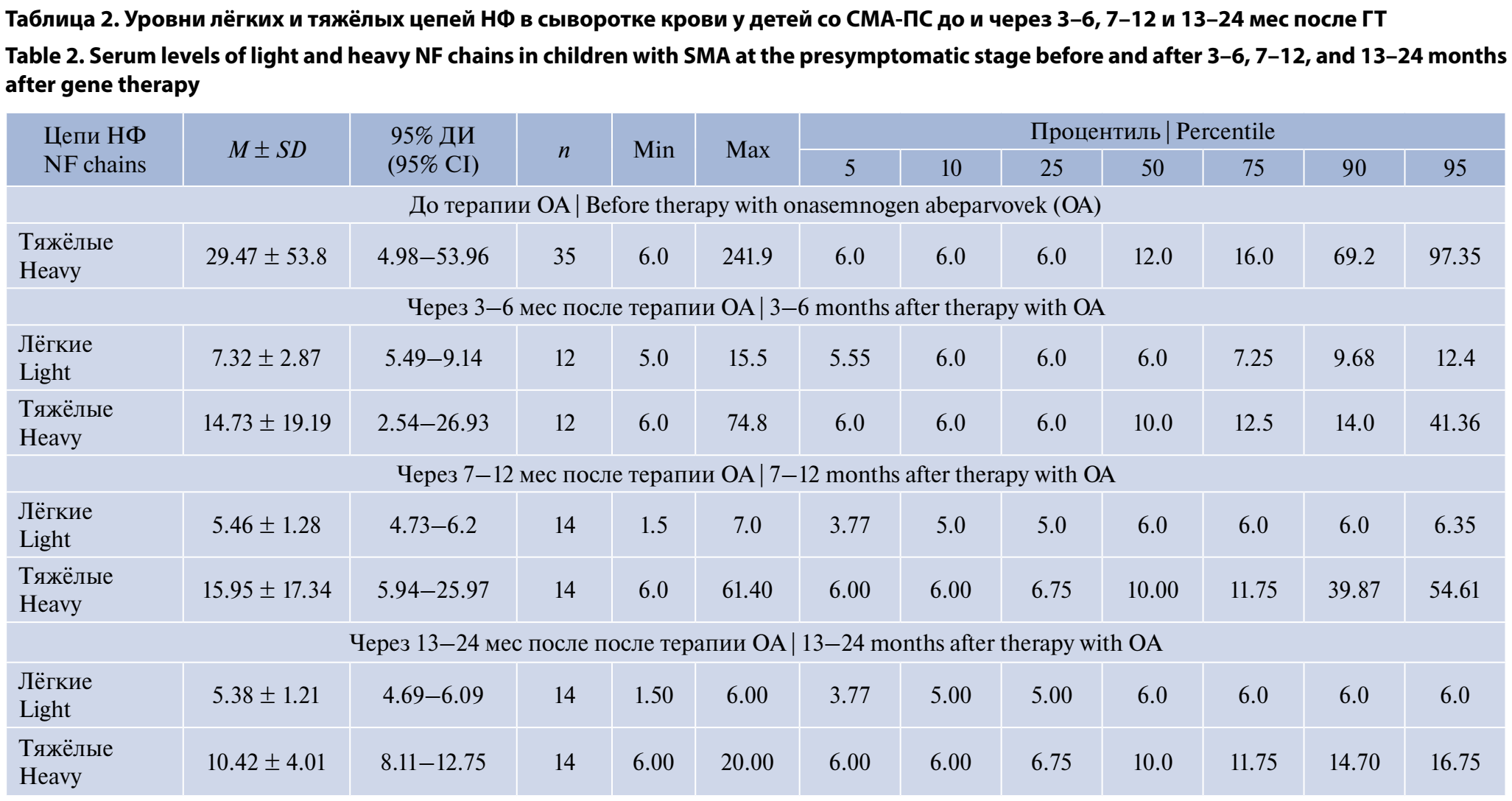
Был проведён сравнительный анализ уровней лёгких и тяжёлых цепей НФ в сыворотке крови пациентов со СМА-ПС в динамике через 3–6, 7–12 и 13–24 мес после ГТ. Отмечено достоверное (р < 0,01) снижение уровней лёгких цепей НФ в сыворотке крови через 3–6 мес после ГТ (6,0 [6,00; 7,25]) по сравнению с уровнями лёгких цепей НФ до лечения (6,0 [6,00; 31,43]) за счёт уменьшения интерквартильных размахов при одинаковом уровне медианы (рис. 3). Через 7–12 и 13–24 мес после ГТ продолжалось снижение интерквартильных размахов при сохранении прежнего уровня медианы. Через 3–6 мес после введения ОА было выявлено снижение уровней тяжёлых цепей НФ и в дальнейшем, через 7–12 и 13–24 мес, — плавное снижение медианы при уменьшении интерквартильных размахов.
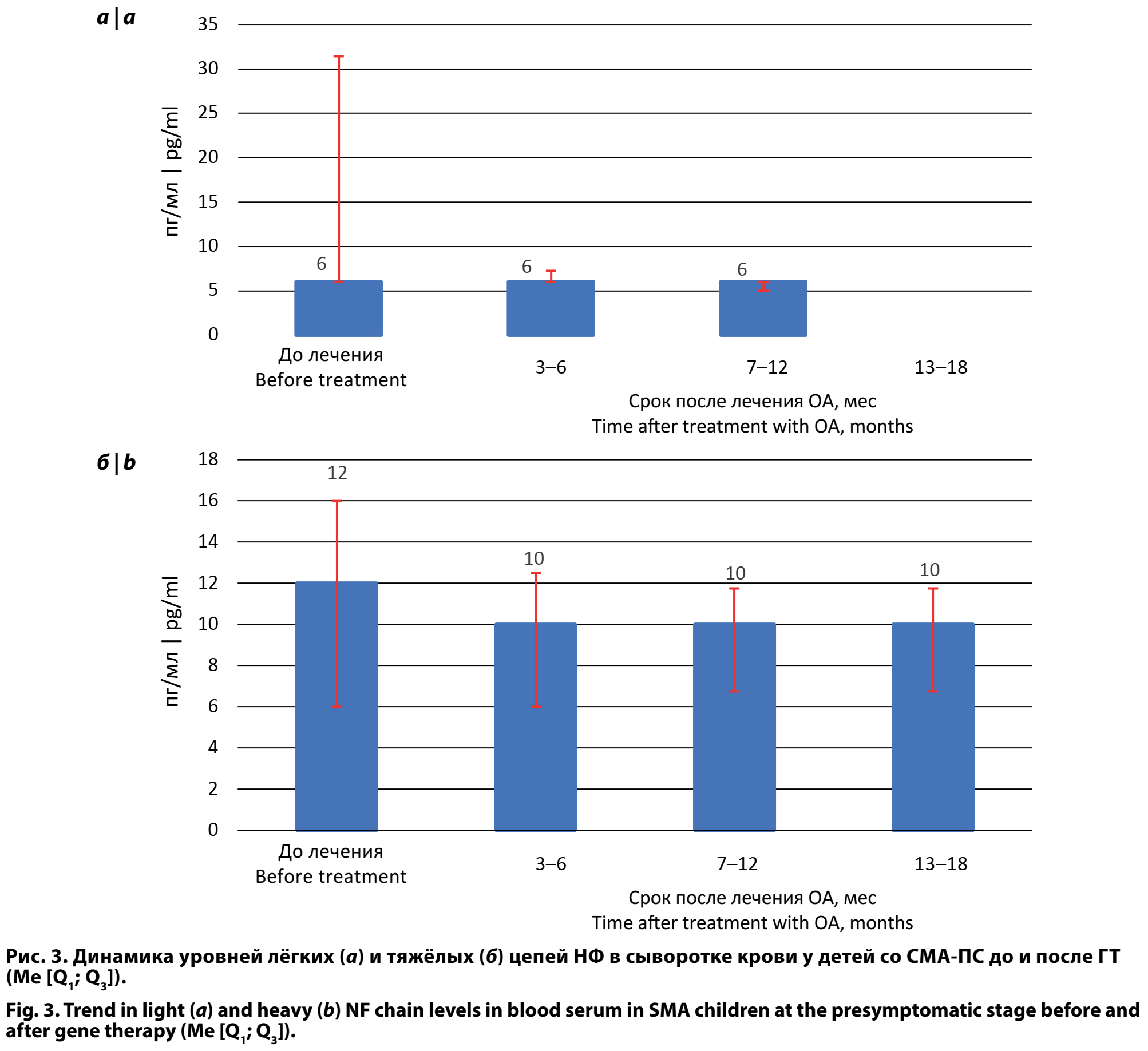
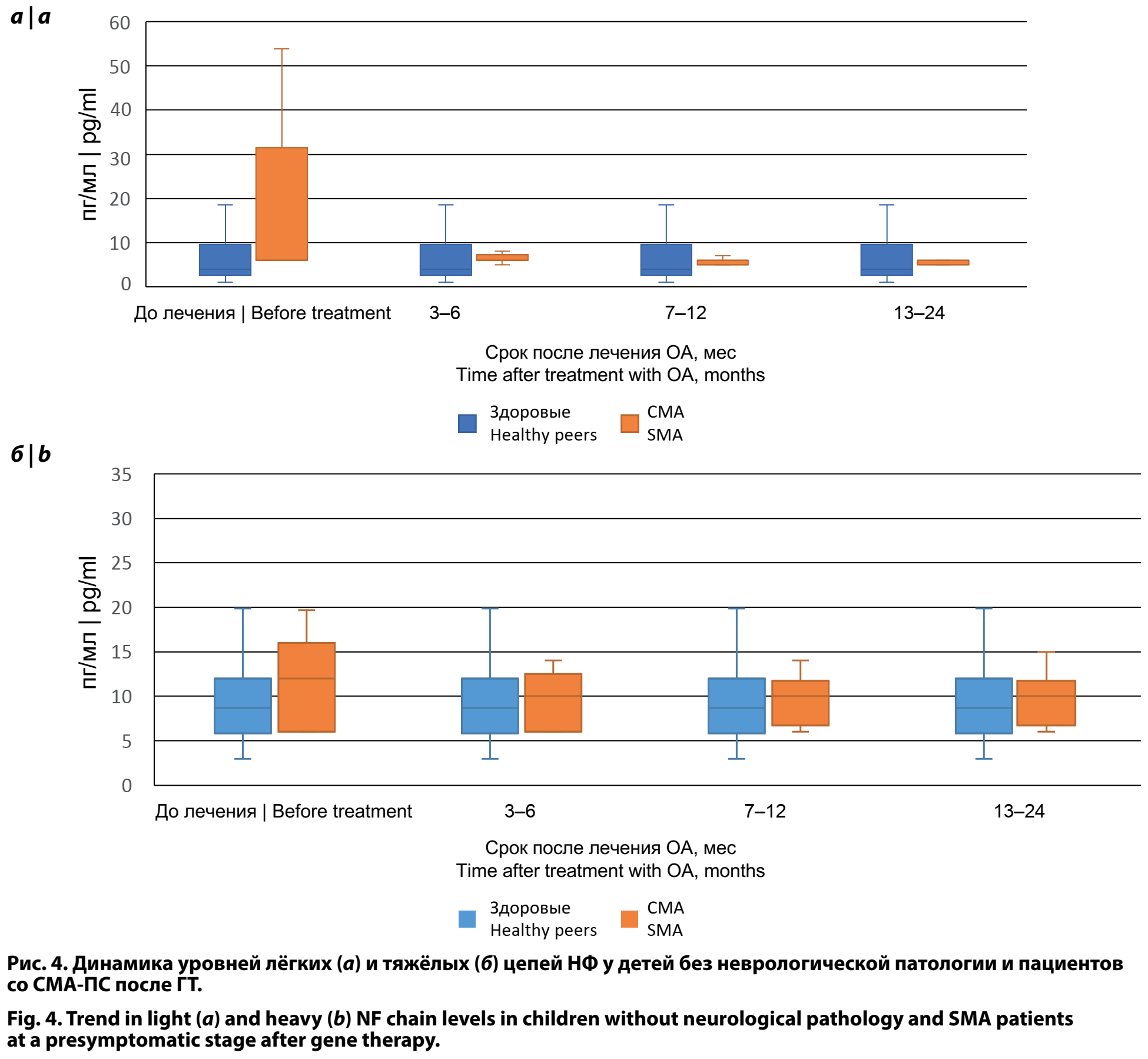
При проведении сравнительного анализа уровней лёгких и тяжёлых цепей НФ в сыворотке крови пациентов со СМА-ПС до ГТ и 76 неврологически здоровых детей в возрасте 0–24 мес (данные получены в исследовании [8]) показано, что уровень лёгких цепей НФ при СМА-ПС был достоверно выше — 6,00 [6,00; 31,43] и 4,00 [2,50; 9,57] пг/мл (p < 0,001). Уровень тяжёлых цепей НФ при сравнении пациентов со СМА-ПС до ГТ со здоровыми сверстниками достоверно не отличался: 12,00 [6,00; 16,00] и 8,70 [5,80; 12,00] пг/мл (p > 0,05).
Сравнительный анализ уровней лёгких и тяжёлых цепей НФ в сыворотке крови между пациентами со СМА-ПС и здоровыми детьми аналогичного возраста в динамике через 3–6, 7–12 и 13–24 мес после ГТ не выявил достоверных различий, что свидетельствует о замедлении процессов нейродегенерации либо о полном их прекращении (рис. 4).
Обсуждение
Проведение патогенетической терапии любого наследственного заболевания на досимптомной стадии наиболее эффективно. Включение СМА в программу неонатального скрининга в России позволило для части пациентов начать патогенетическую, в том числе генную, терапию ещё на ПС заболевания. В связи с этим целью нашего исследования было оценить эффективность ГТ у детей со СМА-ПС на основе изучения моторного развития, определения параметров ЭНМГ и уровней лёгких и тяжёлых цепей НФ в сыворотке крови как до начала лечения, так и в рамках 2-летнего катамнестического наблюдения после лечения.
Дети со СМА-ПС на момент ГТ ОА не имели клинических симптомов болезни, средний возраст составил 2,00 ± 0,94 мес (95% ДИ 1,68–2,32). Необходимо подчеркнуть, что проведённое нами исследование представляет собой наиболее крупный массив данных из одного Центра, продолжительностью до 24 мес наблюдения в катамнезе.
У пациентов со СМА-ПС на фоне ГТ максимальное количество баллов по шкале CHOP-INTEND было достигнуто к 6 мес жизни, по шкале HINE-2 — к 13–18 мес жизни, при этом темпы двигательного развития соответствовали нормативным критериям ВОЗ. Данные нашей работы полностью согласуются с результатами ранее проведённых зарубежных исследований.
Так, в исследовании SPR1NT у пациентов со СМА-ПС с 3 копиями гена SMN2 из 15 детей, получавших генную терапию, 14 могли стоять без поддержки и 11 могли ходить самостоятельно по нормативам ВОЗ [9]. При дальнейшем наблюдении было показано, что дети, не достигшие двигательных навыков по возрасту, в более старшем возрасте овладевали этими навыками.
В исследовании SPR1NT все 14 пациентов со СМА-ПС с 2 копиями гена SMN2 овладели навыком самостоятельного сидения и 11 (79%) из них достигли этого двигательного рубежа в пределах нормального периода развития, установленного ВОЗ [10]; 11 (79%) из 14 детей со СМА-ПС могли стоять без опоры и 7 (50%) из них достигли этого навыка в рамках нормального моторного развития по ВОЗ. Десять (71%) из 14 детей ходили самостоятельно и 6 (43%) сделали это в рамках нормативов ВОЗ.
Результаты, опубликованные J.W. Day и соавт., I. Desguerre и соавт., C. Weiß и соавт., свидетельствуют о том, что на фоне проведения патогенетической терапии у детей со СМА-ПС, верифицированных при неонатальном скрининге, как с тремя, так и с двумя копиями гена SMN2 средний возраст достижения первых моторных навыков был меньше по сравнению с пациентами со СМА I типа [11–13]. Кроме того, среди пациентов со СМА-ПС значительно большая доля детей достигала основных этапов двигательного развития в рамках нормативов ВОЗ. Все представленные выше результаты неоспоримо указывают на необходимость раннего выявления заболевания и своевременного лечения пациентов со СMA как с 2, так и с 3 копиями гена SMN2. Исходные показатели и среднее общее увеличение показателей по шкале HINE-2, полученные в исследовании I. Desguerre и соавт., были значительно выше у пациентов со СМА-ПС по сравнению с детьми со СМА I типа.
В исследовании SPR1NT также анализировались пациенты со СМА-ПС с 2 копиями гена SMN2. У таких детей исходная оценка по шкале CHOP-INTEND составляла 46,1 ± 8,8 балла и быстро повышалась после инфузии ОА [10]. Увеличение оценки по шкале CHOP-INTEND через 1 мес после лечения составило 3,9 ± 8,3 балла. В возрасте 3 мес значение по шкале CHOP-INTEND увеличилось на 11,2 ± 8,8 балла и в возрасте 6 мес оценка по шкале CHOP-INTEND составила 60 (51–64) баллов. Все 14 детей со СМА-ПС в исследовании SPR1NT с 2 копиями гена SMN2 достигли показателя CHOP-INTEND, как минимум, 58 баллов к 18 мес [10]. Безусловно, сравнивать напрямую эти данные с результатами нашего исследования нельзя, т. к. у нас преобладали дети со СМА-ПС с 3 копиями гена SMN2, но общая тенденция совпадает.
Таким образом, результаты нашего исследования показали, что применение ГТ ОА у пациентов со СМА-ПС привело к достижению основных этапов двигательного развития в соответствии с нормативами ВОЗ.
При проведении ЭНМГ параметры М-ответа и СРВ в нашей когорте пациентов со СМА-ПС до старта терапии значимо не отличались от нормативных данных, и на фоне лечения ОА было отмечено повышение амплитуды М-ответа и СРВ при увеличении возраста у всех пациентов.
В исследовании NURTURE у пациентов со СМА-ПС, получавших нусинерсен, в течение 1-го года жизни также было отмечено увеличение амплитуды М-ответа, что наблюдалось и в более старшем возрасте. При этом в фоновом обследовании были получены различия между подгруппами пресимптоматических пациентов с 2 и 3 копиями гена SMN2. Исходная средняя амплитуда М-ответа локтевого нерва была выше у пациентов с 3 копиями гена SMN2 (3,11 ± 1,12 мВ) по сравнению с пациентами с 2 копиями (2,69 ± 1,52 мВ) [14]. При катамнестическом наблюдении у детей с 2 копиями гена SMN2 (n = 5) к 1849-му дню после старта терапии препаратом нусинерсен средняя амплитуда М-ответа составила 4,52 ± 2,20 мВ (среднее изменение от исходного уровня — 2,32 ± 1,93 мВ). У детей с 3 копиями гена SMN2 (n = 10) на 1373-й день после начала лечения среднее значение амплитуды М-ответа составило 8,17 ± 2,96 мВ (среднее изменение от исходного уровня — 5,06 ± 3,13 мВ) [15]. Поскольку в нашем исследовании пациенты со СМА-ПС имели в подавляющем большинстве 3 копии гена SMN2, нам не удалось провести статистически значимого сравнения показателей ЭНМГ в зависимости от числа копий гена SMN2.
Параметры M-ответа при СМА хорошо коррелируют с двигательной функцией, и их определение в динамике может иметь потенциальную ценность в качестве инструментального маркера заболевания. Таким образом, определение максимальной амплитуды М-ответа при ЭНМГ является легко выполнимым, валидным и надёжным показателем конечного результата при СМА для использования в клинических исследованиях и реальной практике в педиатрии.
До проведения лечения в нашем исследовании у пациентов со СМА-ПС в сравнении с показателями детей без неврологической патологии достоверно выше был только уровень лёгких цепей НФ в сыворотке крови (p < 0,001). Уже через 3–6 мес после ГТ уровень лёгких цепей НФ в сыворотке крови снижался у детей со СМА-ПС и не отличался от результатов детей без неврологической патологии, что сохранялось при тестировании через 7–12 и 13–24 мес.
Сходная тенденция была отмечена в исследовании пациентов со СМА-ПС при тестировании тяжёлых цепей НФ в сыворотке крови и ликворе на фоне лечения нусинерсеном [14]. Исходные уровни тяжёлых цепей НФ были выше у пациентов с 2 копиями гена SMN2 по сравнению с пациентами с 3 копиями (р = 0,0050 в сыворотке крови и р = 0,0020 в ликворе). Уровень тяжёлых цепей НФ у пациентов со СМА-ПС быстро снижался на фоне лечения нусинерсеном и затем стабилизировался. Аналогичная тенденция значений тяжёлых цепей НФ наблюдалась в ликворе.
Высокие значения уровня НФ в сыворотке крови до лечения и их быстрое снижение на фоне патогенетической терапии детей со СМА следует рассматривать как маркер эффективности терапии, поскольку увеличение уровней НФ происходит уже на ПС, ещё до появления клинической симптоматики и изменений на ЭНМГ.
Ограничения исследования. Наблюдение за пациентами осуществлялось в условиях реальной клинической практики из-за чего часть пациентов не всегда строго придерживалась графика контрольных осмотров. Продолжительность наблюдения не у всех пациентов со СМА-ПС соответствовала 2 годам. Это связано с тем, что неонатальный скрининг на СМА стартовал в России с января 2023 г. и дети на ПС болезни включались по мере их выявления при молекулярно-генетической диагностике. Таким образом, к моменту завершения набора материала в исследование (март 2025 г.) из всех 35 пациентов со СМА-ПС 30 детей находились под наблюдением в течение минимум 12 мес, 23 детей — в течение 18 мес, 12 детей — в течение 24 мес после ГТ ОА.
Заключение
Пациенты со СМА-ПС в нашем исследовании как до, так и после ГТ имели моторное развитие в соответствии с возрастными нормами и достоверно не отличались от детей без неврологической патологии. При сравнительном анализе показателей ЭНМГ детей со СМА и группы здоровых детей установлено, что у пациентов со СМА-ПС на фоне ГТ статистически значимых различий не получено. Применение ОА у пациентов со СМА-ПС приводило к значимому снижению уровня лёгких цепей НФ в сыворотке крови. Максимальное снижение показателей НФ отмечалось через 3–6 мес после ГТ.
Таким образом, применение ГТ на ПС позволило у большинства пациентов предупредить развитие симптомов СМА.
Наши данные подтверждают результаты других исследований о том, что пациенты, выявленные с помощью неонатального скрининга и получившие патогенетическое лечение в раннем возрасте, имеют более высокие исходные значения моторного развития и в целом демонстрируют более значительные улучшения [9, 16, 17].
1. Mercuri E., Sumner C.J., Muntoni F., Darras B.T., Finkel R.S. Spinal muscular atrophy. Nat. Rev. Dis. Primers. 2022; 8(1): 52. https://doi.org/10.1038/s41572-022-00380-8
2. Yeo C.J.J., Tizzano E.F., Darras B.T. Challenges and opportunities in spinal muscular atrophy therapeutics. Lancet Neurol. 2024; 23(2): 205–18. https://doi.org/10.1016/S1474-4422(23)00419-2
3. Schroth M.K., Deans J., Bharucha Goebel D.X., Burnette W.B., Darras B.T., Elsheikh B.H., et al. Spinal muscular atrophy update in best practices: recommendations for treatment considerations. Neurol. Clin. Pract. 2025; 15(1): e200374. https://doi.org/10.1212/CPJ.0000000000200374
4. Al-Zaidy S.A., Kolb S.J., Lowes L., Alfano L.N., Shell R., Church K.R., et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J. Neuromuscul. Dis. 2019; 6(3): 307–17. https://doi.org/10.3233/JND-190403
5. Kirschner J., Bernert G., Butoianu N., De Waele L., Fattal-Valevski A., Haberlova J., et al. 2024 update: European consensus statement on gene therapy for spinal muscular atrophy. Eur. J. Paediatr. Neurol. 2024; 51: 73–8. https://doi.org/10.1016/j.ejpn.2024.06.001
6. Петрухин А.С. Развитие нервной системы у новорожденных и детей раннего возраста. Методика исследования. Синдромы поражения. В кн.: Детская неврология. Том 1. М.: ГЭОТАР-Медиа; 2018: 199–221.
7. Фисенко Д.А., Куренков А.Л., Кузенкова Л.М., Черников В.В., Увакина Е.В., Бурсагова Б.И. и др. Нормативные показатели стимуляционной электромиографии у детей раннего возраста. Неврологический журнал имени Л.О. Бадаляна. 2023; 4(4): 193–9. https://doi.org/10.46563/2686-8997-2023-4-4-193-199 https://elibrary.ru/bawhuc
8. Фисенко Д.А., Кузенкова Л.М., Куренков А.Л., Семикина Е.Л., Увакина Е.В., Черников В.В. и др. Динамика уровней лёгких и тяжёлых цепей нейрофиламентов в сыворотке крови детей со спинальной мышечной атрофией на фоне применения генной терапии. Неврологический журнал имени Л.О. Бадаляна. 2025; 6(1): 26–36. https://doi.org/10.46563/2686-8997-2025-6-1-26-36 https://elibrary.ru/usswzn
9. Strauss K.A., Farrar M.A., Muntoni F., Saito K., Mendell J.R., Servais L., et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat. Med. 2022; 28(7): 1390–7. https://doi.org/10.1038/s41591-022-01867-3
10. Strauss K.A., Farrar M.A., Muntoni F., Saito K., Mendell J.R., Servais L., et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat. Med. 2022; 28(7): 1381–9. https://doi.org/10.1038/s41591-022-01866-4
11. Day J.W., Finkel R.S., Chiriboga C.A., Connolly A.M., Crawford T.O., Darras B.T., et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021; 20(4): 284–93. https://doi.org/10.1016/S1474-4422(21)00001-6
12. Desguerre I., Barrois R., Audic F., Barnerias C., Chabrol B., Davion J.B., et al. Real-world multidisciplinary outcomes of onasemnogene abeparvovec monotherapy in patients with spinal muscular atrophy type 1: experience of the French cohort in the first three years of treatment. Orphanet. J. Rare Dis. 2024; 19(1): 344. https://doi.org/10.1186/s13023-024-03326-3
13. Weiß C., Becker L.L., Friese J., Blaschek A., Hahn A., Illsinger S., et al. Efficacy and safety of gene therapy with onasemnogene abeparvovec in children with spinal muscular atrophy in the D-A-CH-region: a population-based observational study. Lancet Reg. Health Eur. 2024; 47: 101092. https://doi.org/10.1016/j.lanepe.2024.101092
14. De Vivo D.C., Bertini E., Swoboda K.J., Hwu W.L., Crawford T.O., Finkel R.S., et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019; 29(11): 842–56. https://doi.org/10.1016/j.nmd.2019.09.007
15. Crawford T.O., Swoboda K.J., De Vivo D.C., Bertini E., Hwu W.L., Finkel R.S., et al. Continued benefit of nusinersen initiated in the presymptomatic stage of spinal muscular atrophy: 5-year update of the NURTURE study. Muscle Nerve. 2023; 68(2): 157–70. https://doi.org/10.1002/mus.27853
16. Servais L., Day J.W., De Vivo D.C., Kirschner J., Mercuri E., Muntoni F., et al. Real-world outcomes in patients with spinal muscular atrophy treated with Onasemnogene Abeparvovec monotherapy: findings from the RESTORE registry. J. Neuromuscul. Dis. 2024; 11(2): 425–42. https://doi.org/10.3233/JND-230122
17. Kariyawasam D.S., D’Silva A.M., Sampaio H., Briggs N., Herbert K., Wiley V., et al. Newborn screening for spinal muscular atrophy in Australia: a non-randomised cohort study. Lancet Child Adolesc. Health. 2023; 7(3): 159–70. https://doi.org/10.1016/S2352-4642(22)00342-X
Доктор мед. наук, профессор, начальник центра детской психоневрологии, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия; Клинический институт детского здоровья имени Н.Ф. Филатова ФГАОУ ВО «Первый МГМУ имени И.М. Сеченова» Минздрава России (Сеченовский университет), 119435, Москва, Россия
Доктор мед. наук, зав. лаб. нервных болезней, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, зав. отделением психоневрологии и нейрореабилитации, заместитель директора по научной работе, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: fisenko.daria@mail.ru
Канд. мед. наук, зав. отделением диагностики и восстановительного лечения, начальник методического аккредитационно-симуляционного центра, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Доктор мед. наук, зав. лабораторным отделом, гл. науч. сотр., врач лабораторной диагностики, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, мл. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, врач-невролог, ст. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, врач-невролог, ст. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, мл. науч. сотр. лаб. детских редких наследственных болезней ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Доктор мед. наук, первый зам. директора, и.о. руководителя Института подготовки медицинских кадров, врач-нефролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Кузенкова Л.М., Куренков А.Л., Увакина Е.В., Фисенко Д.А., Черников В.В., Семикина Е.Л., Попович С.Г., Бурсагова Б.И., Глоба О.В., Андреенко Н.В., Абдуллаева Л.М., Курова Ю.А., Адалимова Н.С., Николенко Д.С., Лялина А.А., Комарова О.В. Результаты двухлетнего комплексного наблюдения за пациентами со спинальной мышечной атрофией, получившими генную терапию препаратом онасемноген абепарвовек на пресимптоматической стадии заболевания. Неврологический журнал имени Л.О. Бадаляна. 2025;6(4):182-194. https://doi.org/10.46563/2686-8997-2025-6-4-182-194. EDN: bletts
Kuzenkova L.M., Kurenkov A.L., Uvakina E.V., Fisenko D.A., Chernikov V.V., Semikina E.L., Popovich S.G., Bursagova B.I., Globa O.V., Andreenko N.V., Abdullaeva L.M., Kurova J.A., Adalimova N.S., Nikolenko D.S., Lyalina A.A., Komarova O.V. Results of a two-year comprehensive follow-up in patients with spinal muscular atrophy who received gene therapy with onasemnogen abeparvovec at the presymptomatic stage of the disease. L.O. Badalyan Neurological Journal. 2025;6(4):182-194. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-4-182-194. EDN: bletts

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















