



Содержание
Перейти к:
https://doi.org/10.46563/2686-8997-2025-6-3-140-152
EDN: lsnkiq
Перейти к:
Введение. Атаксический детский церебральный паралич (аДЦП) — наименее распространённая среди всех форм ДЦП, встречается реже, чем у 1 из 10 пациентов с ДЦП. К возрасту 5 лет более чем у половины из этих детей диагноз пересматривается, и подтверждаются другие состояния, не связанные с ДЦП. Накапливаются данные о том, что фенотипическое сходство с аДЦП может быть у ряда генетических заболеваний. Некоторые из них проявляются с рождения, другие манифестируют позже и имеют прогрессирующее течение.
Цель исследования — изучить особенности клинической картины, лабораторных и инструментальных данных, позволяющих отличить пациентов с аДЦП от пациентов с наследственными формами атаксий.
Материалы и методы. В исследование были включены 59 детей в возрасте от 1 года до 3 лет 8 мес, которые были распределены на 3 группы: с подтверждённым аДЦП (n = 29), наследственными заболеваниями (атаксия с подтверждённым генетическим диагнозом) (n = 13), прогрессирующей атаксией предположительно генетической природы, окончательно не верифицированной (n = 17). Всем детям проводились детальная оценка анамнеза, исследование неврологического статуса, нейровизуализация (МРТ). Выраженность двигательных нарушений оценивали с помощью шкалы GMFSC, а степень тяжести атаксии — по разработанной нами Педиатрической шкале атаксии (ПША). При исследовании с помощью ПША проводилась оценка в баллах развития моторных навыков и симптомов, указывающих на поражение структур ЦНС, отвечающих за координацию движений. По общей сумме баллов тяжесть атаксии рассматривали как лёгкую (1–8 баллов), среднюю (9–13 баллов) и тяжёлую (14–23 баллов). Все пациенты наблюдались в динамике на протяжении 5 лет.
Результаты. При включении в исследование между группами отсутствовали значимые различия средних балльных оценок по ПША, и у большинства пациентов определялась атаксия средней тяжести. Однако динамика оценок по ПША за 5-летний период наблюдения пациентов оказалась разнонаправленной: у детей с аДЦП они имели тенденцию к стабилизации и постепенному улучшению, тогда как для пациентов групп с наследственными и предположительно генетическими атаксиями характерным было их неуклонное ухудшение в связи с нарастанием двигательных расстройств. Следует подчеркнуть сходство динамики показателей по ПША у пациентов двух последних групп.
По данным МРТ, в группе аДЦП преобладали изменения, типичные для гипоксически-ишемического поражения в виде перивентрикулярной лейкопатии (73,3%). Другой часто выявляемой патологией была гипоплазия мозжечка (46,4%). В группе наследственно обусловленной атаксии перивентрикулярная лейкопатия встречалась несколько реже (61,5%), другими находками были корковая атрофия (30,7%), гипомиелинизация (7,6%), гипоплазия ствола мозга (7,6%) и атрофия мозжечка (7,6%).
Заключение. Основными дифференциально-диагностическими критериями между наследственно обусловленными атаксиями и аДЦП являются прогрессирование клинической картины болезни, нарастание изменений на МРТ и молекулярно-генетическое выявление мутаций, которые вызывают заболевания, сопровождающиеся атаксией в детском возрасте.
Соблюдение этических стандартов. Исследование проводилось в соответствии с принципами Хельсинкской декларации, одобрено ЛЭК ФГАОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России (выписка из протокола заседания № 180 от 17.12.2028). Все пациенты или их законные представители подписали добровольное информированное согласие.
Участие авторов:
Ражева Д.С. — сбор и анализ данных, обзор публикаций по теме статьи, написание текста рукописи;
Хондкарян Г.Ш — анализ данных, написание текста статьи, окончательное утверждение для публикации рукописи;
Заваденко Н.Н. — разработка концепции, написание текста статьи, окончательное утверждение для публикации рукописи.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Финансирование. Исследование не имело спонсорской поддержки.
Поступила 01.09.2025
Принята к печати 22.09.2025
Опубликована 31.10.202
Ражева Д.С., Хондкарян Г.Ш., Заваденко Н.Н. Наследственные атаксии, протекающие под маской детского церебрального паралича. Неврологический журнал имени Л.О. Бадаляна. 2025;6(3):140-152. https://doi.org/10.46563/2686-8997-2025-6-3-140-152. EDN: lsnkiq
Razheva D.S., Khondkarian G.Sh., Zavadenko N.N. Наследственные атаксии, протекающие под маской детского церебрального паралича. L.O. Badalyan Neurological Journal. 2025;6(3):140-152. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-3-140-152. EDN: lsnkiq
Введение
Атаксический детский церебральный паралич (аДЦП) — наименее распространённая среди всех форм ДЦП, встречается реже, чем у 1 из 10 пациентов с ДЦП [1–6]. Кроме того, к возрасту 5 лет более чем у половины из этих детей диагноз пересматривается и подтверждаются состояния, не связанные с ДЦП, что указывает на ошибочность первоначального диагноза [1, 7, 8]. Во многих случаях атаксия с ранним началом бывает признаком генетически детерминированных заболеваний с прогрессирующим течением.
Клинический интерес к изучению мозжечковых атаксий в детском возрасте появился после работ Николауса Фридрейха, который опубликовал свою первую статью о «семейной спинно-мозжечковой дегенерации» в 1861 г. и описал её новые случаи в 1863 и 1876 гг. [9]. В последующие десятилетия другие авторы также наблюдали подобных пациентов, и «болезнь Фридрейха» стала считаться отдельной нозологической единицей.
Работы Н. Фридрейха усилили интерес к различным формам атаксии в детском возрасте. Были представлены пациенты, у которых неврологическое расстройство проявлялось с рождения и не было прогрессирующим. Такие истории болезни были обобщены и изучены в конце XIX в. Зигмундом Фрейдом, занимавшимся в то время ДЦП. Он заключил, что эти случаи не имели отношения к болезни Фридрейха, а представляли собой особую форму врождённого церебрального паралича. В 1897 г. З. Фрейд впервые предложил специальную категорию «атаксический церебральный паралич» [9].
Исследования в этом направлении продолжил Фредерик Баттен, который в 1903 г. описал 8 детей с врождённой мозжечковой атаксией, названной им «церебеллярной диплегией» [10]. Лишь у 3 из них в анамнезе имелись указания на патологические роды. Клиническими проявлениями были замедленное двигательное развитие, нарушения координации движений и речи, тремор, у всех пациентов, кроме одного, — мышечная гипотония, у некоторых — нистагм. Ф. Баттен подчеркнул, что с возрастом состояние пациентов с атаксией улучшалось, и они начинали самостоятельно передвигаться без поддержки. Он полагал, что врождённая атаксия может быть результатом повреждения мозжечка во время родов, а в некоторых случаях — пороков развития головного мозга. Баттен рассматривал её как отдельное заболевание, отличное от атаксии Фридрейха [8, 10]. Он также отмечал, что атаксия в детском возрасте может быть приобретённой в результате энцефалопатий, осложняющих инфекционные заболевания, а также внутричерепных абсцессов и опухолей.
В 1909 г. немецкий невролог О. Фёрстер при описании 4 случаев мозжечковой атаксии отметил, что их клинические проявления несколько отличались от ранее описанных, и назвал их примерами «атонически-астатического ДЦП», который следует разграничивать от врождённой мозжечковой атаксии [9]. О. Фёрстер предположил, что это состояние не было связано исключительно с поражением мозжечка, поскольку в 2 похожих случаях при аутопсии был обнаружен атрофический склероз лобных долей. Он также считал, что существуют переходные синдромы между «атонически-астатическим ДЦП» и «церебральной диплегией», поскольку у некоторых пациентов с выраженными нарушениями координации движений конечностей наблюдались повышение мышечного тонуса, усиление сухожильных рефлексов и патологические разгибательные подошвенные рефлексы.
Значительный вклад в изучение атаксической формы ДЦП внесли Л.О. Бадалян и соавт. [11]. Предлагая классифицировать формы ДЦП для раннего и старшего возраста, они обозначили гипотоническую форму ДЦП, «поскольку выраженная мышечная гипотония у детей первого года жизни является основным симптомом формирующихся в дальнейшем атактической и атонически-астатической форм» [11, с. 15]. Обращая внимание на то, что на различиях между этими формами акцентировал внимание ещё О. Фёрстер, они охарактеризовали их следующие клинические особенности: «При атонически-астатической форме резко затруднено принятие вертикальной позы, невозможно сохранение положения тела в пространстве, выражена мышечная гипотония вследствие дефекта системы постурального контроля. Эти нарушения, как правило, сопровождаются тяжёлой умственной отсталостью и задержкой речевого развития» [11, с. 15]. При атактической форме способность удерживать позу нарушена нерезко. Со временем дети обучаются сидеть, стоять и ходить. В клинической картине доминируют расстройства координации движений. Мышечная гипотония умеренная. Психические и речевые нарушения выражены не столь грубо, как при атонически-астатической форме, а иногда интеллект нормальный» [11, с. 15]. Кроме того, Л.О. Бадалян и соавт. «пришли к заключению о необходимости включения в классификацию ряда смешанных форм, таких как спастико-атактическая, спастико-гиперкинетическая, атактико-гиперкинетическая» [11]. В современных классификациях и публикациях представлена единая атаксическая форма ДЦП, но история изучения этого состояния и клиническая практика свидетельствуют о вариабельности клинических проявлений внутри данной группы пациентов.
ДЦП определяется международными экспертами как группа стабильных, непрогрессирующих нарушений развития моторики и поддержания позы, возникших вследствие непрогрессирующего повреждения и/или аномалии развивающегося головного мозга у плода или новорождённого ребёнка, которые приводят к ограничению функциональной активности [1–3, 12]. В пользу диагноза ДЦП свидетельствуют указания на воздействие патологического фактора в анте-, интра- или постнатальном периодах, нарушения моторного развития и двигательной функции, непрогрессирующие структурные изменения ЦНС при нейровизуализации; часто встречаются сопутствующие нарушения (сенсорные, когнитивные, речевые, поведенческие, симптоматическая эпилепсия) и вторичные ортопедические осложнения [1–3, 12, 13].
аДЦП характеризуется нарушением координации мышечной деятельности, что проявляется в снижении точности, силы и ритма движений. Типичными признаками являются туловищная и локомоторная атаксия, дисметрия, интенционный тремор и мышечная гипотония [1–6]. Особенностью аДЦП является относительно редкое выявление структурных изменений головного мозга при нейровизуализации: более чем у половины пациентов МРТ остаётся в пределах нормы, у 30–40% выявляется гипоплазия мозжечка разной степени выраженности [5, 14].
Накапливаются данные о том, что фенотипическим сходством с аДЦП может обладать ряд генетических заболеваний [3, 5, 8, 15–17]. Некоторые из них проявляются с рождения, другие манифестируют позже и имеют прогрессирующее течение.
Цель исследования — выявление особенностей клинической картины, лабораторных и инструментальных данных, позволяющих отличить пациентов с аДЦП от пациентов с наследственными формами атаксий.
Материалы и методы
Критериями диагноза аДЦП и включения в исследование были:
Всем детям проводилась детальная оценка анамнеза: анализ течения беременности, родов, становления двигательных, психических и речевых функций, текущего неврологического и соматического статуса. Для оценки психомоторного развития у детей до 6 лет использовался Денверский скрининговый тест [18]. Выраженность двигательных нарушений оценивали с помощью шкалы GMFSC [19], степень тяжести атаксии — по разработанной нами Педиатрической шкале атаксии (ПША) (табл. 1).
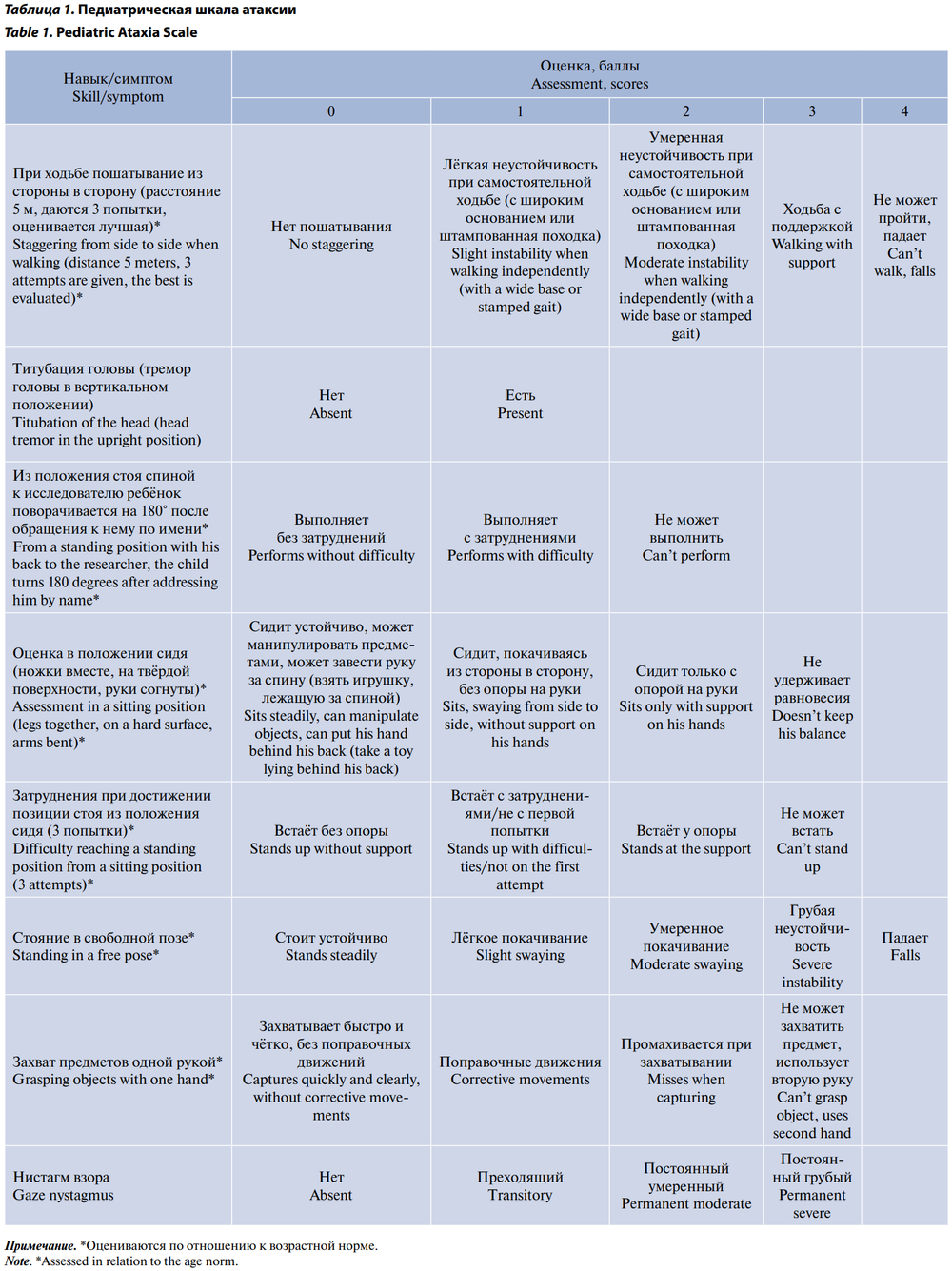
ПША была создана на основании опыта использования Международной объединённой шкалы оценки атаксии ICARS [20] и шкалы оценки двигательной функции и тяжести опсоклонус-миоклонус синдрома [21]. При исследовании с помощью ПША проводили оценку в баллах развития моторных навыков и симптомов, указывающих на поражение структур ЦНС, отвечающих за координацию движений (табл. 1), по общей сумме баллов выраженность клинических проявлений атаксии классифицировали как лёгкую (1–8 баллов), среднюю (9–13 баллов) и тяжёлую (14–23 баллов).
Лабораторное исследование включало общий и биохимический анализы крови, исследование ацилкарнитинов и аминокислот методом тандемной масс-спектрометрии, газохроматографический анализ мочи. Проводили МРТ головного мозга, электроэнцефалографию, по показаниям — электронейромиографию (стимуляционную и игольчатую). Пациенты наблюдались у специалистов (офтальмолога, генетика), а также направлялись на молекулярно-генетическое обследование, объём которого определял врачом-генетик.
Всего в исследование были включены 59 пациентов (36 мужского и 23 женского пола) в возрасте от 1 года до 3 лет 8 мес с первоначальным направительным диагнозом аДЦП. Все пациенты наблюдались нами в динамике на протяжении 5 лет.
Результаты
По данным, полученным в ходе комплексного обследования и динамического наблюдения, 59 пациентов были распределены на 3 группы (табл. 2): с подтверждённым аДЦП (n = 29), наследственными заболеваниями (атаксия с подтверждённым генетическим диагнозом) (n = 13), с прогрессирующей атаксией предположительно генетической природы, окончательно не верифицированной (n = 17).
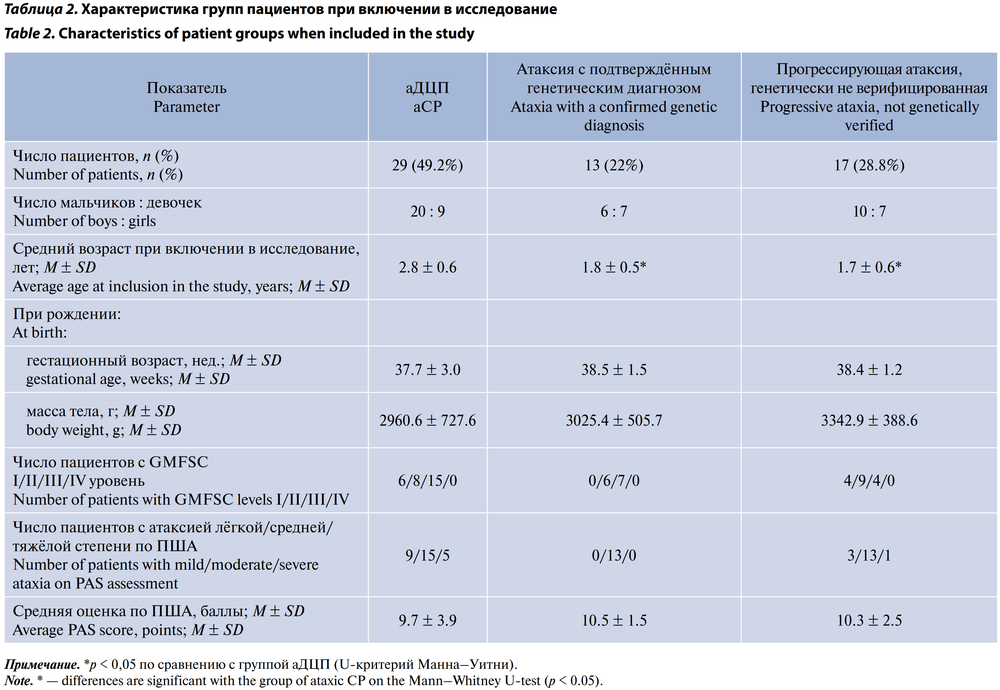
Средний возраст на момент включения в исследование был несколько выше в группе аДЦП (2,8 года), что представляется логичным, т. к. окончательный диагноз ДЦП чаще выставляется позже, после попыток самостоятельной ходьбы. В группах наследственной и предположительно генетической атаксии средний возраст был ниже (1,8 и 1,7 года соответственно), что может быть связано с ранней манифестацией выраженной неврологической симптоматики и её дальнейшим нарастанием. При этом пациенты 3 групп существенно не различались по гестационному возрасту и массе тела при рождении.
По уровням GMFSC (двигательная активность) в группе аДЦП наблюдалось больше тяжёлых форм (III уровень — 15 из 29), в то время как в группе наследственной атаксии не было пациентов с лёгкими формами (GMFSC I отсутствует), и у большинства пациентов уровень GMFSC соответствовал II–III (табл. 2). Это показывает, что выраженные двигательные нарушения характерны как для аДЦП, так и для наследственных атаксий, но отсутствие лёгких форм в генетической группе может служить дополнительным диагностическим ориентиром.
Исследование раннего анамнеза показало, что в группе пациентов с аДЦП чаще регистрировались факторы перинатального риска (патология течения беременности и родов) — всего в 93,1% случаев (табл. 3), в частности, фетоплацентарная недостаточность (17,2%), преждевременные роды (13,8%), экстренное кесарево сечение (31%).
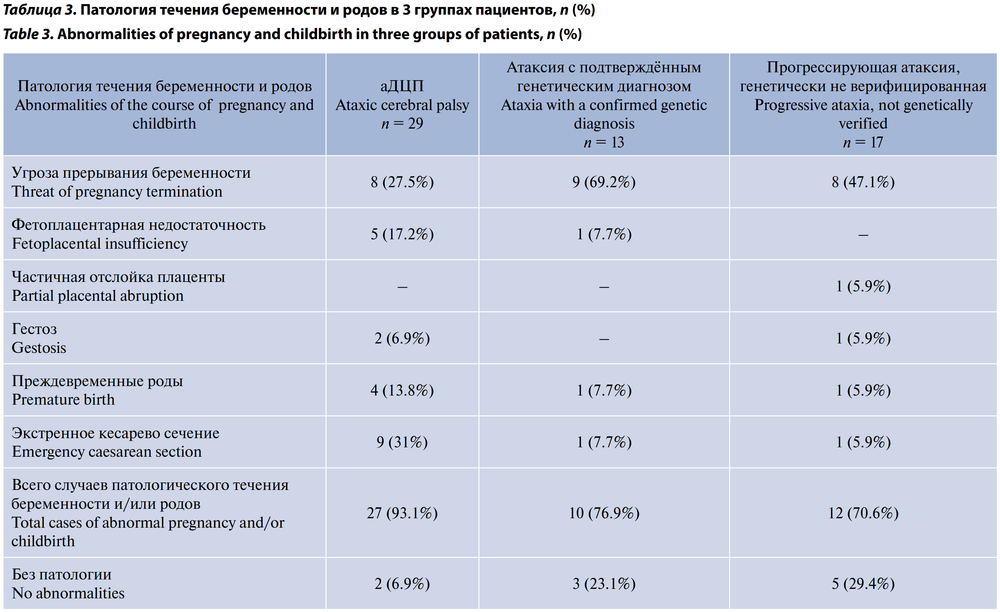
В группах пациентов с наследственно обусловленной и предположительно генетической атаксией патологическое течение беременности и родов отмечалось несколько реже — в 76,9 и 70,6% случаев соответственно. Это подтверждает, что у пациентов с атаксией при отсутствии значимых указаний на отягощённый перинатальный анамнез необходима настороженность в отношении возможных генетических заболеваний.
С другой стороны, угроза прерывания беременности чаще встречалась в анамнезе пациентов с атаксией с подтверждённым (69,2%) и предполагаемым (47,1%) диагнозом генетического заболевания, чем в группе пациентов с аДЦП (27,5%).
При включении в исследование между группами отсутствовали значимые различия средних балльных оценок по ПША, и у большинства пациентов определялась атаксия средней тяжести. Однако динамика оценок по ПША за 5-летний период наблюдения пациентов оказалась разнонаправленной: у детей с аДЦП они имели тенденцию к стабилизации и постепенному улучшению, тогда как для пациентов групп с наследственными и предположительно генетическими атаксиями характерным было их неуклонное ухудшение в связи с нарастанием двигательных расстройств (табл. 4). Следует подчеркнуть сходство динамики показателей по ПША у пациентов двух последних групп.

Единственным исключением в группе атаксий с подтверждённым генетическим диагнозом стал мальчик в возрасте 3 лет при включении в исследование, у которого предполагался аДЦП, но при генетическом исследовании был подтверждён дефицит транспортёра глюкозы 1-го типа (GLUT1) — болезнь де Виво. Наблюдались следующие клинические проявления: начальные симптомы с 2 мес — глазодвигательные нарушения в виде пароксизмов крупноразмашистых движений глазных яблок в разные стороны, длительность пароксизмов 5–15 мин, частота 3–4 раза в неделю. Постепенно продолжительность этих пароксизмов наросла до 15 мин. Атаксия проявилась в 1 год, в 1 год 1 мес появились билатеральные тонико-клонические судорожные приступы, частота которых составила 1 раз в 6 мес, в связи с чем была назначена терапия вальпроатами. Родители ребёнка отмечали флюктуацию атаксии и глазодвигательных нарушений в течение дня. В 3 года также наблюдалась задержка речевого развития (стал произносить отдельные слова). Постановка диагноза дефицита GLUT1 позволила пересмотреть терапию (назначить кетогенную диету и через год постепенно отменить вальпроаты), а также добиться значительного регресса двигательных нарушений: оценка по ПША с исходной в 9 баллов через год улучшилась до 4 баллов и оставалась на данном уровне за весь период дальнейшего наблюдения.
У 13 (22%) пациентов наследственные заболевания были подтверждены методами генетической диагностики и лабораторными исследованиями. У 12 из них (за исключением 1 пациента с болезнью де Виво) наблюдалось прогрессирующее течение заболевания (табл. 5).
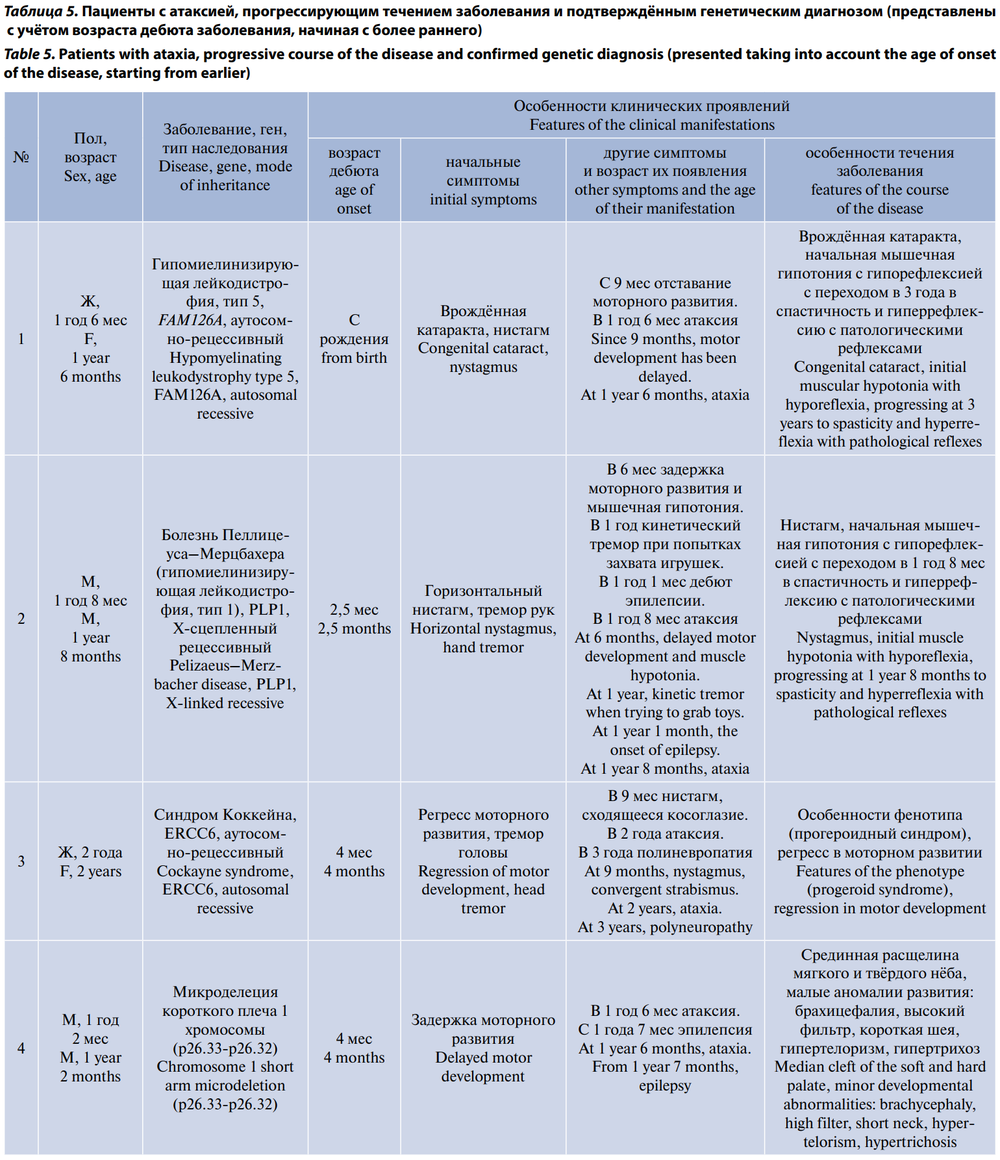
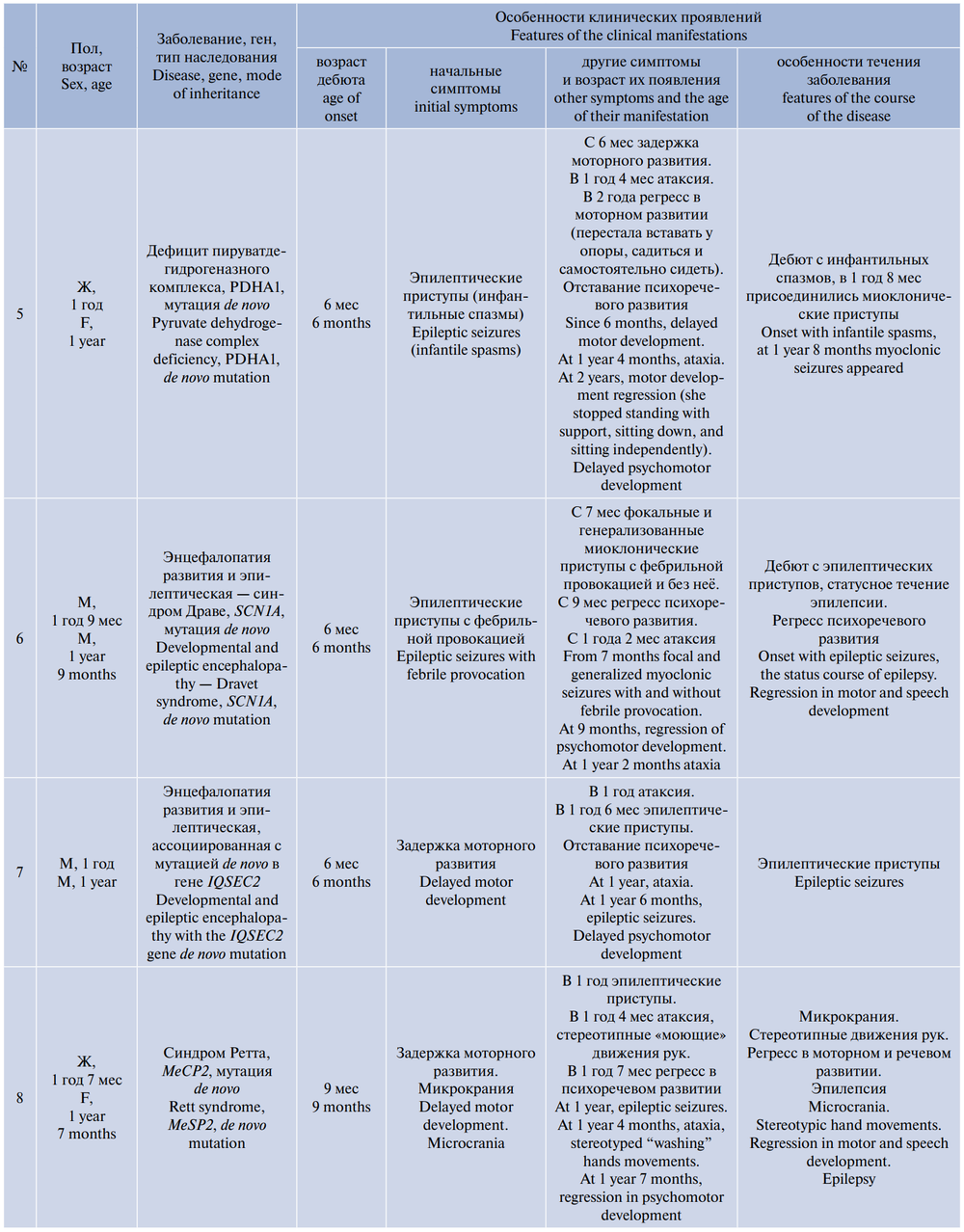
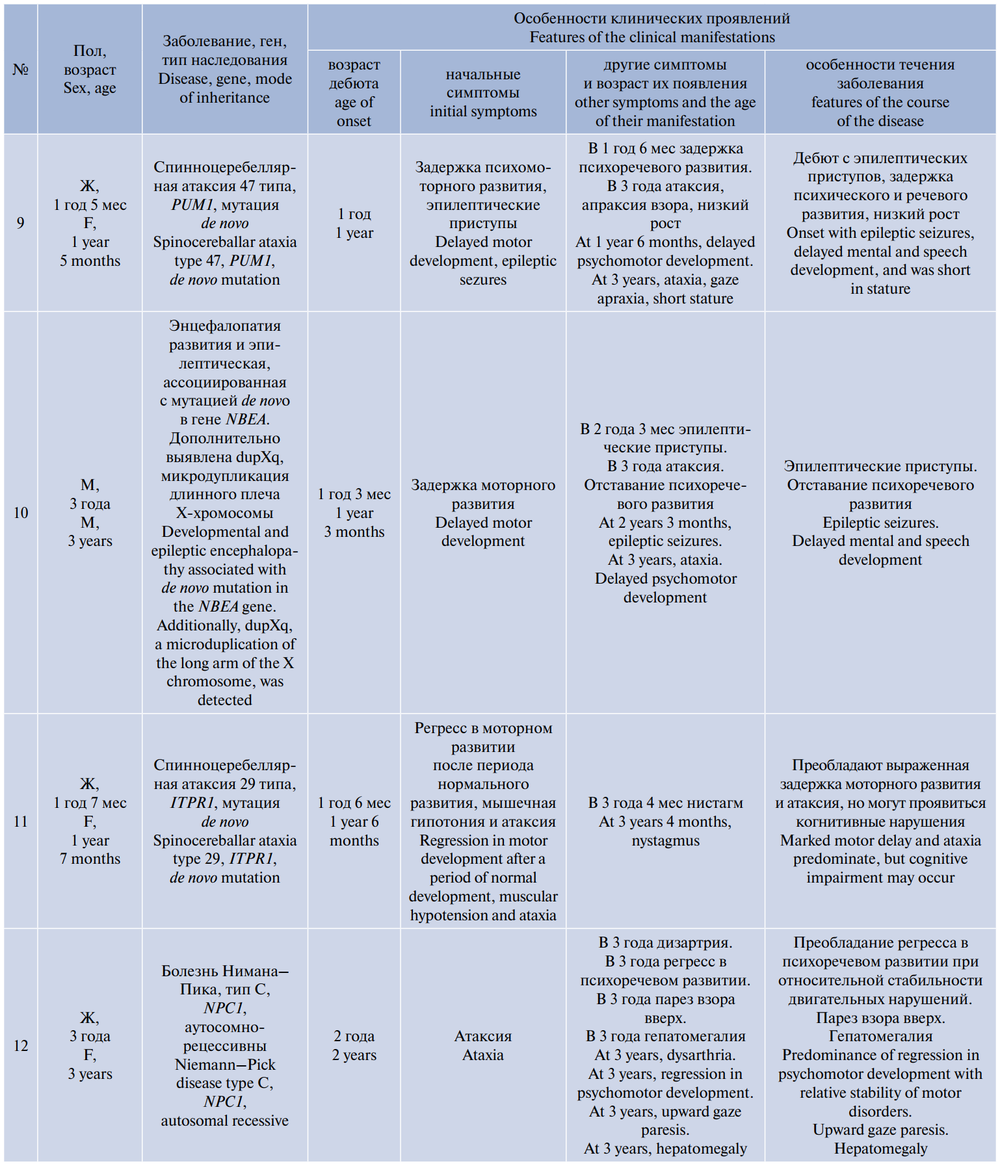
По результатам обследования и динамического наблюдения диагноз ДЦП был пересмотрен ещё у 17 (28,8%) пациентов, у которых отмечалось прогрессирование неврологической симптоматики, в ряде случаев — нарастание изменений на МРТ. Эти пациенты вошли в подгруппу «Прогрессирующая атаксия, генетически не верифицированная». Однако диагностический поиск в этих случаях продолжается, что в перспективе может привести к подтверждению диагнозов генетических заболеваний. Прогрессирующее течение заболевания у этих пациентов проявлялось утратой или ухудшением ранее достигнутых двигательных навыков, регрессом в психическом и речевом развитии, присоединением эпилептических приступов у 3 (17,6%) пациентов, гиперкинезов у 4 (23,5%) пациентов, глазодвигательных нарушений у 4 (23,5%) пациентов. У 3 пациентов по данным нейровизуализации выявлено прогрессирование структурных изменений в головном мозге: увеличение атрофии мозжечка, изменений белого вещества больших полушарий по типу лейкодистрофии, а также развитие субкортикальных кист на фоне атрофических процессов.
Характеристика структурных изменений ЦНС в 3 группах пациентов по данным МРТ представлена в табл. 6.
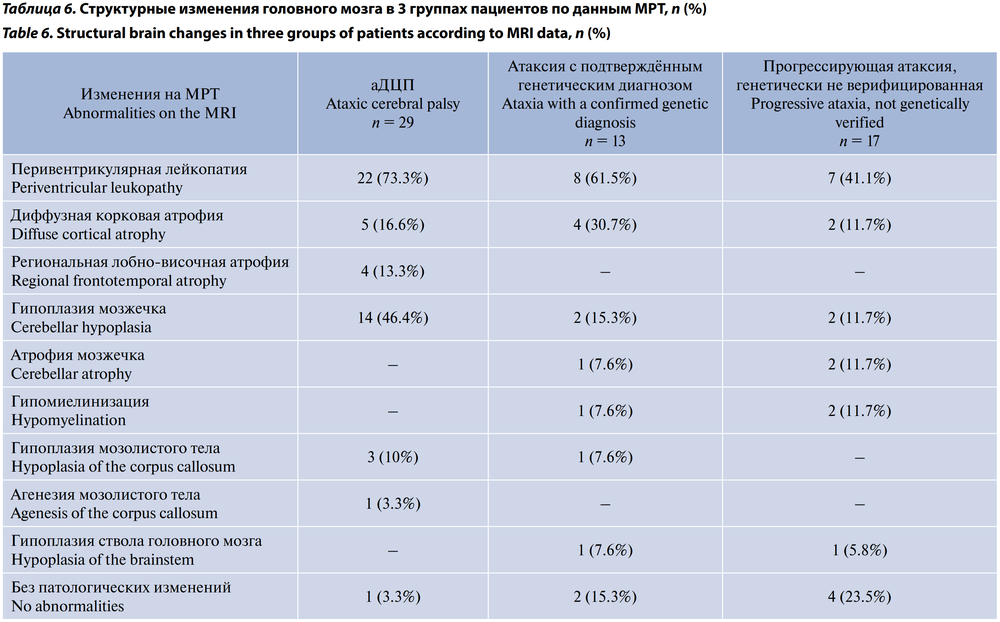
В группе аДЦП преобладали изменения, типичные для гипоксически-ишемического поражения в виде перивентрикулярной лейкопатии (73,3%). Другой часто выявляемой при МРТ патологией была гипоплазия мозжечка (46,4%).
В группе наследственно обусловленной атаксии перивентрикулярная лейкопатия встречалась несколько реже (61,5%), другими находками были корковая атрофия (30,7%), гипомиелинизация (7,6%), гипоплазия ствола мозга (7,6%) и атрофия мозжечка (7,6%).
В группе прогрессирующей атаксии изменения МРТ варьировали: у 41,1% — лейкопатия, у 11,7% — гипомиелинизация и атрофия мозжечка, у 11,7% — корковая атрофия, в 23,5% случаев изменений не было.
Обсуждение
Результаты исследования подтверждают, что диагноз аДЦП часто оказывается «рабочим» и нередко маскирует под собой наследственные заболевания. По данным литературы [3, 5, 7, 14], настораживающими факторами, требующими пересмотра диагноза аДЦП, являются:
Полученные нами результаты в целом согласуются с данными литературы.
Выявление у значительного числа пациентов генетической этиологии заболевания подчёркивает важность проведения молекулярно-генетического анализа даже при наличии факторов риска перинатального поражения мозга. Это объясняется тем, что этиологический диагноз ДЦП устанавливается, исходя из анамнеза и клинических данных, тогда как наследственные заболевания могут манифестировать сходной симптоматикой, особенно у детей раннего возраста, и быть ошибочно отнесены к последствиям гипоксически-ишемического поражения ЦНС. Более того, наличие таких симптомов, как нистагм, парез взора, неаккомодационное косоглазие, эпилептические приступы и гиперкинезы, особенно в сочетании с прогрессирующим ухудшением неврологического статуса, требует от врача-невролога высокой клинической настороженности. Эти признаки не являются типичными для классического течения аДЦП и должны рассматриваться как настораживающие в отношении наследственно-дегенеративных расстройств и указания на необходимость расширенного диагностического поиска, включая консультацию врача-генетика и молекулярно-генетическое исследование.
Мутации могут иметь рецессивный характер или возникать de novo, в связи с чем в семейном анамнезе не обнаруживается других случаев заболевания и складывается впечатление о том, что он не отягощён. Об этом следует помнить неврологу при расспросе родственников больного. Поэтому современным методам генетической диагностики принадлежит важная роль, поскольку они позволяют установить конкретную причину заболевания и своевременно назначить адекватную терапию.
Дополнительным аргументом в пользу пересмотра диагноза ДЦП в ряде случаев является анализ данных нейровизуализации. В группе пациентов с наследственными формами атаксий и прогрессирующей атаксической симптоматикой чаще выявлялись изменения, характерные для диффузных дегенеративных процессов: гипомиелинизация, корковая атрофия, а также признаки лейкодистрофии, что контрастирует с перивентрикулярной лейкопатией вследствие гипоксически-ишемического поражения ЦНС, типичной для ДЦП. Однако даже при отсутствии изменений на МРТ нельзя исключать генетическую природу заболевания: отсутствие морфологических маркёров не означает отсутствие заболевания, особенно в случаях с прогрессирующим клиническим течением.
Тем не менее, по результатам нашего исследования, основными дифференциально-диагностическими критериями между наследственными атаксиями и аДЦП являются прогрессирование клинической картины болезни, нарастание изменений на МРТ и молекулярно-генетическое выявление мутаций, которые вызывают заболевания, сопровождающиеся атаксией в детском возврате. Ни анамнестические данные, включая особенности течения беременности и родов, ни результаты однократных или проведённых с короткими интервалами неврологических обследований, ни характер изменений на МРТ в большинстве случаев не являются абсолютно надёжной информацией для дифференциального диагноза. В перспективе именно на молекулярно-генетическую диагностику, которая становится всё более доступной, в частности на полногеномное или полноэкзомное исследования, всё больше будут опираться специалисты для проведения раннего дифференциального диагноза ДЦП и наследственных атаксий.
Важно подчеркнуть, что ранняя верификация генетической природы заболевания имеет важное практическое значение: установление точного диагноза позволяет прогнозировать течение, определять риск для будущих беременностей в семье, а также, что особенно важно, открывает возможности для проведения таргетной терапии. Некоторые из выявленных состояний, например дефицит пируватдегидрогеназного комплекса или болезнь Нимана–Пика типа С, могут быть частично компенсированы с помощью патогенетической терапии, что существенно влияет на прогноз.
Заключение
Диагностические трудности разграничения между аДЦП и наследственно обусловленными атаксиями требуют комплексного подхода, включающего тщательный анализ данных анамнеза, неврологическое обследование, нейровизуализацию и молекулярно-генетическую диагностику. Результаты настоящего исследования подчёркивают необходимость формирования новых диагностических стандартов: дети с атактическим синдромом и атипичной клинической картиной должны рассматриваться как кандидаты на углублённое генетическое обследование, а подход «диагноз ДЦП на всю жизнь» требует пересмотра.
1. Баранов А.А., Намазова-Баранова Л.С., Кузенкова Л.М., Куренков А.Л., Клочкова О.А. Детский церебральный паралич у детей: Клинические рекомендации. М.; 2016.
2. Батышева Т.Т., Гузева В.И., Куренков А.Л., Змановская В.А., Быкова О.В., Ахадова Л.Я. и др. Детский церебральный паралич. В кн.: Гузева В.И., ред. Федеральное руководство по детской неврологии. СПб.: Валетудо; 2023: 184–214.
3. Немкова С.А., ред. Детский церебральный паралич: руководство для врачей. М.: ГЭОТАР-Медиа; 2025.
4. Levy J.P., Oskoui M., Ng P., Andersen J., Buckley D., Fehlings D., et al. Ataxic-hypotonic cerebral palsy in a cerebral palsy registry: Insights into a distinct subtype. Neurol. Clin. Pract. 2020; 10(2): 131–9. https://doi.org/10.1212/cpj.0000000000000713
5. Horber V., Andersen G.L., Arnaud C., De La Cruz J., Dakovic I., Greitane A., et al. Prevalence, clinical features, neuroimaging, and genetic findings in children with ataxic cerebral palsy in Europe. Neurology. 2023; 101(24): e2509–21. https://doi.org/10.1212/WNL.0000000000207851
6. Pettersson K., Johansen M., Jahnsen R., Rodby-Bousquet E. Characteristics of children with ataxic cerebral palsy. BMC Pediatr. 2025; 25(1): 335. https://doi.org/10.1186/s12887-025-05681-x
7. Chen A., Dyck Holzinger S., Oskoui M., Shevell M. Canadian Cerebral Palsy Registry. Losing a diagnosis of cerebral palsy: a comparison of variables at 2 and 5 years. Dev. Med. Child Neurol. 2020; 62(1): 83–8. https://doi.org/10.1111/dmcn.14309
8. Dan B. How useful is the diagnosis of ataxic cerebral palsy? Dev. Med. Child Neurol. 2020; 62(3): 264. https://doi.org/10.1111/dmcn.14453
9. Ingram T.T.S. Congenital ataxic syndromes in cerebral palsy. Acta Paediatrica. 1962; 51(2): 209–21. https://doi.org/10.1111/j.1651-2227.1962.tb06531.x
10. Batten F.E. Congenital cerebellar ataxia (cerebellar diplegia). Clinical. J. 1903; 22: 81–8.
11. Бадалян Л.О., Журба Л.Т., Тимонина О.В. Детские церебральные параличи. Киев: Здоровье; 1988.
12. Bax M., Goldstein M., Rosenbaum P., Leviton A., Paneth N., Dan B., et al. Executive Committee for the Definition of Cerebral Palsy. Proposed definition and classification of cerebral palsy, April 2005. Dev. Med. Child Neurol. 2005; 47(8): 571–6. https://doi.org/10.1017/s001216220500112x
13. Пак Л.А., Кузенкова Л.М., Фисенко А.П., Куренков А.Л. Детский церебральный паралич: клинические и инструментальные характеристики. Российский педиатрический журнал. 2019; 22(1): 4–11. https://elibrary.ru/zqxznj
14. Korzeniewski S.J., Birbeck G., DeLano M.C., Potchen M.J., Paneth N. A systematic review of neuroimaging for cerebral palsy. J. Child Neurol. 2008; 23(2): 216–27. https://doi.org/10.1177/0883073807307983
15. Eskandar M., Tochen L., Shin M.R., Lavenstein B., Meltzer M., Gropman A., et al. Limitations of multigene next-generation sequencing panel for “cerebral palsy” phenotype and other complex movement disorders. Pediatr. Neurol. 2023; 149: 15–8. https://doi.org/10.1016/j.pediatrneurol.2023.08.040
16. Jang D.H., Kim J., Schwabe A.L., Lotze T.E. Genetics of cerebral palsy: diagnosis, differential diagnosis, and beyond. Ann. Rehabil. Med. 2024; 48(6): 369–76. https://doi.org/10.5535/arm.240081
17. Pearson T.S., Pons R., Ghaoui R., Sue C.M. Genetic mimics of cerebral palsy. Mov. Disord. 2019; 34(5): 625–36. https://doi.org/10.1002/mds.27655
18. Frankenburg W.K., Dodds J., Archer P., Shapiro H., Bresnick B. The Denver II: a major revision and restandardization of the Denver Developmental Screening Test. Pediatrics. 1992; 89(1): 91–7.
19. Palisano R., Rosenbaum P., Walter S., Russell D., Wood E., Galuppi B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev. Med. Child Neurol. 1997; 39(4): 214–23. https://doi.org/10.1111/j.1469-8749.1997.tb07414.x
20. Schoch B., Regel J.P., Frings M., Gerwig M., Maschke M., Neuhäuser M., et al. Reliability and validity of ICARS in focal cerebellar lesions. Mov. Disord. 2007; 22(15): 2162–9. https://doi.org/10.1002/mds.21543
21. Rossor T., Yeh E.A., Khakoo Y., Angelini P., Hemingway C., Irani S.R., et al. OMS Study Group. Diagnosis and management of opsoclonus-myoclonus-ataxia syndrome in children: an international perspective. Neurol. Neuroimmunol. Neuroinflamm. 2022; 9(3): e1153. https://doi.org/10.1212/NXI.0000000000001153
Ассистент кафедры неврологии, нейрохирургии и медицинской генетики им. Л.О. Бадаляна Института нейронаук и нейротехнологий ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России, 117513, Москва, Россия
e-mail: darvezhar@mail.ru
Доктор мед. наук, профессор кафедры неврологии, нейрохирургии и медицинской генетики им. Л.О. Бадаляна Института нейронаук и нейротехнологий ФГАОУ ВО РНИМУ им Н.И. Пирогова Минздрава России, 117513, Москва, Россия
e-mail: gareguin@mail.ru
Доктор мед. наук, профессор, зав. кафедрой неврологии, нейрохирургии и медицинской генетики им. Л.О. Бадаляна Института нейронаук и нейротехнологий ФГАОУ ВО РНИМУ им Н.И. Пирогова Минздрава России, 117513, Москва, Россия
e-mail: zavadenko@mail.ru
Ражева Д.С., Хондкарян Г.Ш., Заваденко Н.Н. Наследственные атаксии, протекающие под маской детского церебрального паралича. Неврологический журнал имени Л.О. Бадаляна. 2025;6(3):140-152. https://doi.org/10.46563/2686-8997-2025-6-3-140-152. EDN: lsnkiq
Razheva D.S., Khondkarian G.Sh., Zavadenko N.N. Наследственные атаксии, протекающие под маской детского церебрального паралича. L.O. Badalyan Neurological Journal. 2025;6(3):140-152. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-3-140-152. EDN: lsnkiq

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















