



Содержание
Перейти к:
https://doi.org/10.46563/2686-8997-2025-6-3-133-139
EDN: bdbbqm
Перейти к:
Введение. Цель исследования — оценка динамики двигательной активности с помощью сбора анамнестических данных и динамического наблюдения за пациентами с миодистрофией Дюшенна, вызванной нонсенс-мутацией (нмМДД), в Московской области на фоне патогенетической терапии препаратом аталурен.
Материалы и методы. Динамическое наблюдение 13 пациентов с нмМДД осуществляли на базе Центра детской психоневрологии Научно-исследовательского клинического института детства Московской области. В данном исследовании проводился ретроспективный анализ анамнестических данных, двигательной активности пациентов по тестам «Северная Звезда» (NSAA) и 6-минутной ходьбы (6MWT). Для оценки эффективности терапии препаратом аталурен было проведено непрямое сравнение с данными из регистров STRIDE и CINRG DNHS.
Результаты. В исследуемой группе пациентов возраст начала самостоятельной ходьбы составил 15,7 ± 4,5 мес, возраст установки диагноза — 60,2 ± 21,7 мес (5,0 ± 1,8 года). 92% пациентов (n = 12) получают глюкокортикостероиды (ГКС), 5 из них — преднизолон, 7 — дефлазакорт. Средний возраст начала приёма ГКС составил 7,1 ± 0,9 года. Возраст инициации патогенетической терапии препаратом аталурен составил 7,0 ± 1,3 года. Длительность наблюдения пациентов варьировалась от 1,5 до 8 лет. Данные по пациентам с верифицированными результатами спирометрии (46%) и эхокардиографии (92%) за последние 12 мес распределились следующим образом: снижения фракции выброса левого желудочка не зарегистрировано ни у одного пациента, снижение форсированной жизненной ёмкости лёгких лёгкой степени наблюдалось у 1 пациента. Регулярные реабилитационные мероприятия получают 8 (62%) пациентов. Для оценки двигательных функций использовали тесты NSAA (n = 10) и 6MWT (n = 8). При анализе данных с помощью 6MWT наблюдалось увеличение пройденного расстояния у 6 (75%) пациентов, уменьшение — у 2 (25%). Результаты теста NSAA: у 2 (20%) пациентов отмечается улучшение, у 6 (60%) — стабилизация состояния, у 2 (20%) — ухудшение (в том числе у 1 — потеря амбулаторности).
Заключение. Своевременная диагностика и лечение пациентов в соответствии с клиническими рекомендациями играют ключевую роль в успешной терапии пациентов с МДД. Патогенетическая терапия препаратом аталурен замедляет скорость прогрессирования заболевания и улучшает выживаемость пациентов с нмМДД.
Соблюдение этических стандартов. Законные представители пациентов дали добровольное информированное согласие на обработку персональных данных.
Участие авторов:
Нахушева Ф.И. — концепция и дизайн статьи, написание текста, редактирование;
Серов А.В. — концепция и дизайн статьи, написание текста, редактирование;
Лапочкин О.Л. — редактирование.
Все соавторы — утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Конфликт интересов. Нахушева Ф.И., Лапочкин О.Л. являются лекторами, получающими гонорары от фармацевтической компании ООО «ПиТиСи Терапьютикс».
Поступила: 20.08.2025
Принята к печати: 10.09.2025
Опубликована: 31.10.2025
Нахушева Ф.И., Серов А.В., Лапочкин О.Л. Опыт терапии пациентов с миодистрофией Дюшенна, вызванной нонсенс-мутацией. Неврологический журнал имени Л.О. Бадаляна. 2025;6(3):133-139. https://doi.org/10.46563/2686-8997-2025-6-3-133-139. EDN: bdbbqm
Nakhusheva F.I., Serov A.V., Lapochkin O.L. The experience of treating patients with Duchenne myodystrophy caused by nonsense mutation. L.O. Badalyan Neurological Journal. 2025;6(3):133-139. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-3-133-139. EDN: bdbbqm
Введение
Прогрессирующая мышечная дистрофия Дюшенна (МДД) представляет собой редкое наследственное Х-сцепленное нервно-мышечное заболевание, обусловленное мутациями в гене DMD. Это заболевание было впервые описано французским врачом Гаспаром Дюшенном в 1861 г. и является наиболее распространённой формой мышечной дистрофии у детей [1]. Распространённость МДД варьируется в зависимости от популяции, но в среднем составляет около 1 на 3500 новорождённых мальчиков [2]. Примерно в 8% случаев проявления МДД могут также наблюдаться у лиц женского пола, являющихся носителем дефектного гена [3]. По оценкам, в мире насчитывается более 250 тыс. пациентов с МДД [4].
Как правило, мутация в гене DMD является наследственной, т. е. передаётся от матери, однако в трети случаев она возникает de novo. Спектр мутаций достаточно широкий: примерно в 65% случаев встречаются крупные делеции; около 10% мутаций представлены дупликациями, а остальные случаи — точковыми и малыми мутациями, из которых 10–15% составляют нонсенс-мутации (стоп-мутации) [5]. Нонсенс-мутации приводят к появлению преждевременного стоп-кодона в мРНК, что нарушает процесс транскрипции и приводит к образованию укороченного нефункционального белка дистрофина.
Ген DMD состоит из 79 экзонов и кодирует белок дистрофин, который играет ключевую роль в поддержании структуры и функции мышечных клеток скелетных мышц, дыхательной и сердечно-сосудистой систем [6, 7]. Дистрофин обеспечивает связь между цитоскелетом мышечных клеток и базальной мембраной, что критически важно для их стабильности и защиты от механических повреждений. В ответ на повреждение мышечных клеток активируются воспалительные процессы, что усугубляет дегенерацию тканей. Со временем происходит замещение мышц жировой и соединительной тканью, и этот процесс прогрессирует на протяжении всей жизни пациента [8].
Характерные симптомы МДД включают прогрессирующую мышечную слабость, особенно в проксимальных отделах, а также повышенную утомляемость. Дети с этим заболеванием демонстрируют характерную «утиную» походку и приёмы Говерса («вставание лесенкой»). Также наблюдаются псевдогипертрофия икроножных мышц, ходьба на пальцах ног, «крыловидные» лопатки, гиперлордоз в поясничном отделе позвоночника, снижение сухожильных рефлексов и тонуса мышц [8, 9].
Наиболее заметные проявления начинаются в возрасте 3–5 лет, когда у детей возникают трудности в повседневных физических задачах, таких как бег и ходьба по лестнице. В среднем к 8–10 годам пациенты теряют возможность самостоятельно передвигаться и становятся неамбулаторными больными. Смерть обычно наступает вследствие кардиореспираторных осложнений (дилатационная кардиомиопатия, нарушения проводимости, аритмии, дыхательная недостаточность) в среднем к 20–30 годам [8].
Важно отметить, что на всех стадиях заболевания наблюдается выраженное повышение уровня креатинфосфокиназы (зачастую в сотни раз), что является одним из главных маркеров для диагностики МДД.
Независимо от типа мутации пациентам назначается терапия глюкокортикостероидами (ГКС) системного действия. Данную терапию назначают до начала прогрессирующего ухудшения двигательных функций, когда естественного развития моторных функций у ребенка больше не отмечается [8]. ГКС оказывают положительный эффект за счёт противовоспалительного действия: они замедляют утрату мышечной силы и функции, уменьшают риск развития ортопедических осложнений и стабилизируют функциональное состояние дыхательной и сердечно-сосудистой систем [10, 11]. Исследования продемонстрировали, что применение ГКС продлевает амбулаторное состояние пациентов с МДД в среднем на 3 года [12].
Важную роль в терапии пациентов с МДД играет повседневный двигательный режим с избеганием чрезмерных нагрузок и экцентрических упражнений с высоким сопротивлением [13], а также регулярные курсы реабилитационных мероприятий как амбулаторно, так и стационарно [9, 14].
Помимо ГКС, существуют варианты патогенетической терапии при ряде делеций и точковых нонсенс-мутациях в гене DMD, способные замедлить прогрессирование заболевания [15].
Аталурен — первый препарат для лечения МДД, вызванной нонсенс-мутацией в гене дистрофина (нмМДД), у пациентов старше 2 лет с возможностью самостоятельного передвижения. Препарат был зарегистрирован в России в 2020 г. и включён в клинические рекомендации по МДД [16]. Аталурен относится к классу препаратов «читающих через стоп-кодон» агентов [17]. Он взаимодействует с рибосомой и способствует игнорированию преждевременного стоп-кодона во время трансляции мРНК. Это позволяет рибосоме продолжить синтез белка до естественного стоп-кодона, восстанавливая производство полноразмерного функционального белка. Важно отметить, что аталурен не влияет на нормальные стоп-кодоны в конце гена, что снижает риск образования нефункциональных или токсичных белков [18, 19].
Цель исследования — с помощью сбора анамнестических данных и динамического наблюдения за пациентами с нмМДД в Московской области оценить динамику двигательной активности на фоне патогенетической терапии аталуреном.
Материалы и методы
Динамическое наблюдение пациентов с МДД по Московской области осуществляется на базе Центра детской психоневрологии Научно-исследовательского клинического института детства Московской области. За период работы центра с 2021 г. наблюдаются и беспрерывно получают терапию аталуреном в дозе 40 мг/кг в сутки за счёт средств государственного фонда «Круг добра» 13 пациентов. У всех мальчиков имеется подтверждённая секвенированием по Сенгеру нонсенс-мутация в гене DMD.
В данном исследовании мы провели ретроспективный анализ анамнестических данных, двигательной активности пациентов по шкалам для оценки эффективности терапии аталуреном у пациентов в Московской области, а также проанализировали безопасность проводимой терапии.
Анамнестические данные включали следующие показатели:
Для оценки двигательных функций использовали тесты:
Для оценки эффективности терапии аталуреном мы провели непрямое сравнение с данными из регистров:
Результаты
В исследуемой группе пациентов (n = 13) возраст начала самостоятельной ходьбы составил 15,7 ± 4,5 мес, возраст установки диагноза — 60,2 ± 21,7 мес (5,0 ± 1,8 года) (таблица).
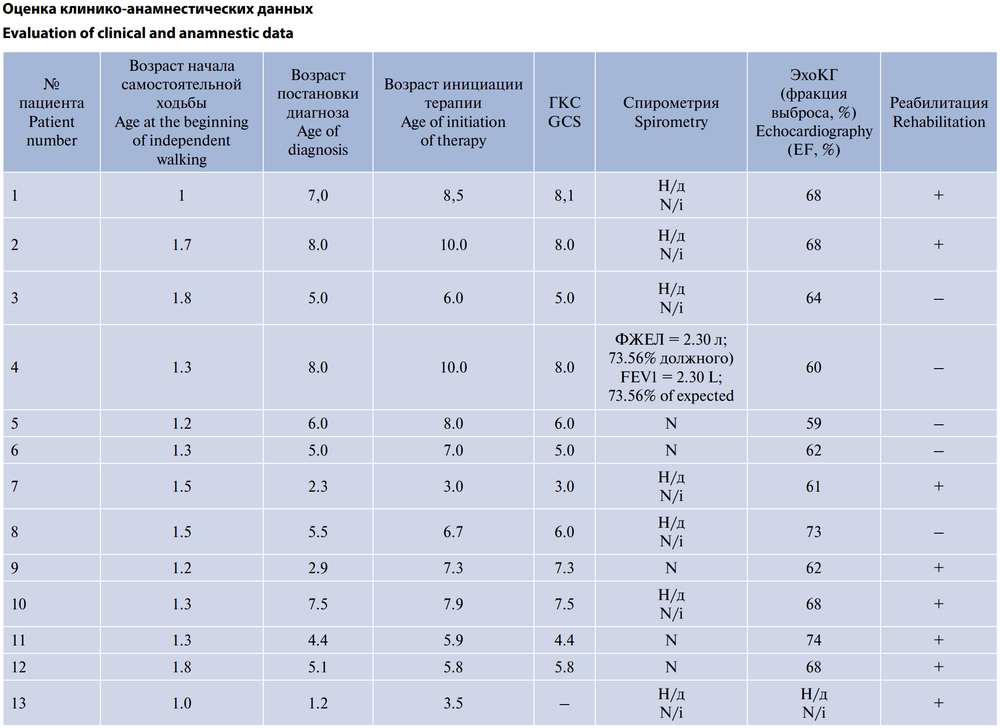
92% пациентов (n = 12) получают ГКС: 5 — преднизолон, 7 — дефлазакорт. Средний возраст начала приёма ГКС составил 7,1 ± 0,9 года, однако следует отметить, что 67% пациентов начали приём ГКС в течение 6 мес после установления диагноза.
Возраст инициации патогенетической терапии препаратом аталурен составил 7,0 ± 1,3 года (самое раннее начало — 3 года, самое позднее — 10 лет). Длительность наблюдения пациентов варьирует от 1,5 до 8 лет.
Данные по пациентам с верифицированными результатами спирометрии (46%) и ЭхоКГ (92%) за последние 12 мес распределились следующим образом: снижения ФВ ЛЖ не зарегистрировано ни у одного пациента, снижение ФЖЕЛ лёгкой степени наблюдается у 1 пациента.
Регулярные реабилитационные мероприятия по данным медицинской документации и со слов родителей получают 8 (62%) пациентов.
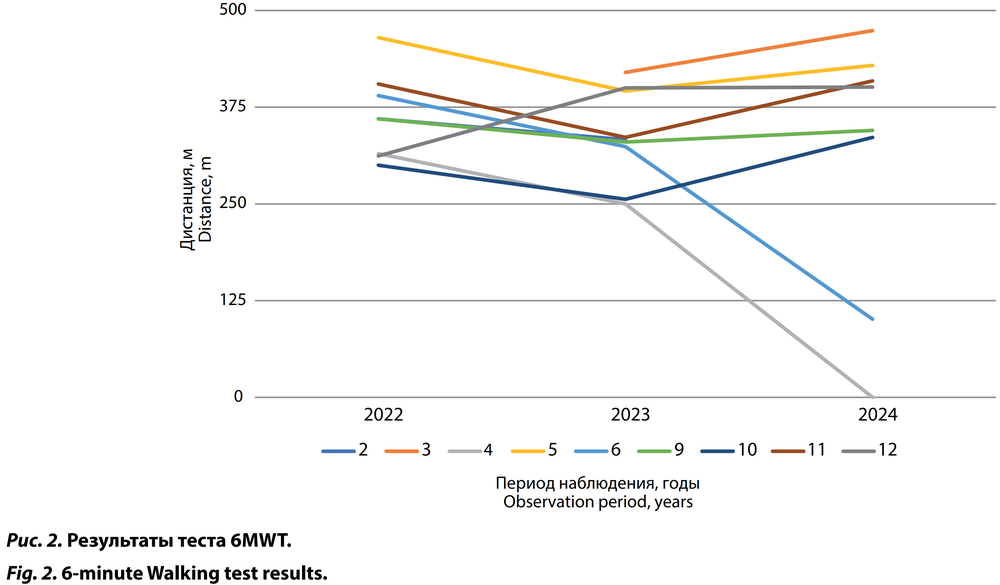
Для оценки двигательных функций использовали тесты NSAA и 6MWT. Из-за задержки психоречевого развития и нарушения поведения не удалось провести тестирование по NSAA 3 пациентам, по 6MWT — 5.
При анализе данных с помощью 6MWT увеличение пройденного расстояния установлено у 6 (75%) пациентов, уменьшение — у 2 (25%) (рис. 1).
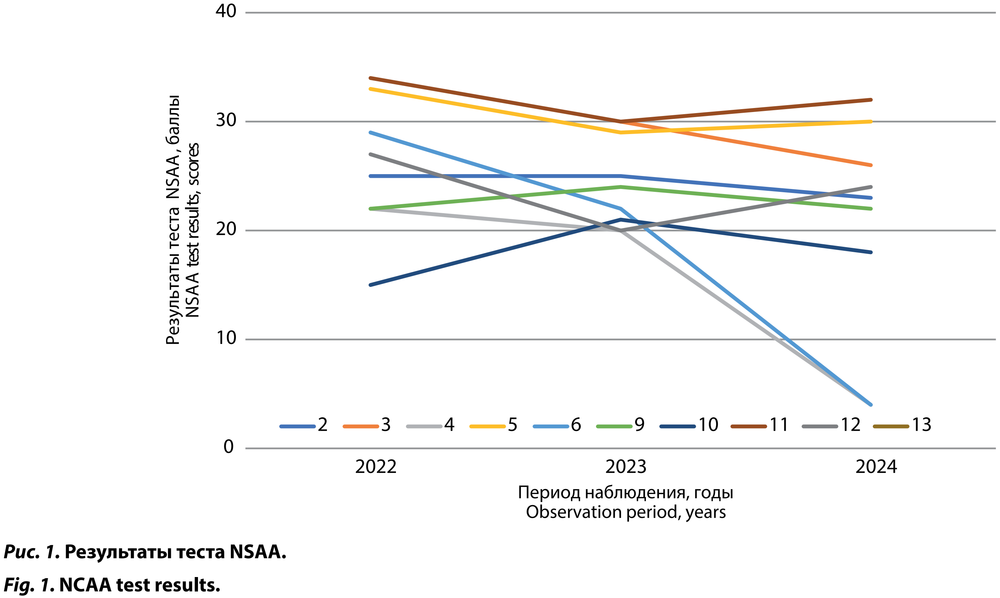
Результаты теста NSAA оценивали согласно исследованию V. Ricotti и соавт., в соответствии с которым при естественном течении заболевания наблюдается снижение на 8 баллов в год после 7 лет [20]. В нашем исследовании у 2 (20%) пациентов отмечено улучшение состояния, у 6 (60%) — стабилизация, у 2 (20%) — ухудшение (в том числе у 1 — потеря амбулаторности) (рис. 2).
Обсуждение
У пациентов с нмМДД в Московской области средний возраст начала самостоятельной ходьбы составил 15 мес, что коррелирует с данными исследований MD STAR и PPMD [21].
Заболевание было диагностировано у наших пациентов в среднем в 5 лет. Несмотря на то, что это соответствует результатам исследования E. Ciafaloni и соавт. [22], у некоторых пациентов наблюдалась выраженная задержка в диагностике (самое позднее — 8 лет), что обусловлено неспецифическими ранними признаками заболевания и возможным отсутствием настороженности у врачей и родителей по отношению к нервно-мышечным патологиям. Однако возраст инициации терапии аталуреном составил 7 лет, что несколько меньше, чем в исследовании STRIDE (8,9 года) [19].
Почти все пациенты (92%) получают терапию ГКС, из них 67% начали приём в течение 6 мес после диагностики заболевания, а средний возраст составил 7 лет, что коррелирует с данными исследования STRIDE [19]. В различных исследованиях было показано, что ГКС увеличивают возраст потери амбулаторности на 2,1–4,4 года и возраст потери функции верхних конечностей на 2,8–8,0 лет [12, 23]. Наиболее оптимальной схемой терапии является ежедневный приём 0,75 мг/кг преднизолона в сутки или 0,90 мг/кг дефлазакорта в сутки [24]. Однако терапия ГКС связана с побочными эффектами, такими как повышение массы тела, задержка роста, остеопороз и т. д., что, вероятно, объясняет небольшую задержку с назначением данной группы препаратов [16].
Анализ результатов ЭхоКГ и спирометрии за последние 12 мес показал сохранение нормальной систолической функции левого желудочка у всех пациентов и снижение ФЖЕЛ лёгкой степени только у 17-летнего пациента, на основании чего мы можем косвенно говорить об эффективности проводимой терапии аталуреном.
Стоит отметить связь кардиомиопатии и снижения функции лёгких с клинической стадией заболевания, особенно заметны данные изменения у пациентов после потери амбулаторности [25]. Патологии сердца и/или лёгких являются сильным маркёром последующей высокой смертности, в частности, 5-летняя выживаемость после падения ФЖЕЛ ниже 1 л составляет всего 8% [26].
Регулярные реабилитационные мероприятия получают только 62% пациентов, что связано в первую очередь с низким уровнем комплаентности родителей и пациентов. Регулярные занятия адаптированной лечебной физкультурой, растяжки по заинтересованным суставам, ношение ортезных изделий позволяют предотвратить мышечную атрофию и развитие вторичных осложнений, таких как контрактуры суставов и сколиотические деформации [9].
В международном наблюдательном исследовании по безопасности и эффективности аталурена (STRIDE) продемонстрировано, что пациенты, получающие патогенетическую терапию аталуреном, значимо дольше сохраняют способность самостоятельно передвигаться (p < 0,0001) по сравнению с пациентами, получающими исключительно стандартное лечение по данным исследования CINRG DNHS. Также, только у 21% пациентов, использовавших аталурен, ФЖЕЛ оказался ниже 60%, в то время как в группе стандартной терапии данный показатель был менее 60% у 36,5% пациентов (p = 0,0021) [19]. Это подчёркивает способность аталурена долго сохранять дыхательную функцию у больных с нмМДД.
Для динамической оценки двигательных функций при МДД используются множество тестов, главными из которых являются тесты NSAA и 6MWT. Проведение тестов может быть затруднено у пациентов младше 5 лет, а также у детей с особенностями когнитивного развития. По данным причинам нам не удалось провести тестирование по NSAA 3 пациентам, по 6MWT — 5 пациентам.
Оценка двигательной функции с помощью теста NSAA в нашем исследовании показала следующие результаты: улучшение на 1–2 балла у 2 пациентов и стабилизацию состояния у 6 пациентов. По NSAA и 6MWT выраженное ухудшение выявлено только у 2 пациентов 14 и 17 лет, а потеря амбулаторности наблюдалась только у 1 пациента в возрасте 17 лет. По данным V. Ricotti и соавт., при естественном течении заболевания регистрируется увеличение баллов по NSAA до 7 лет и снижение на 8 баллов в год после 7 лет, а по данным метаанализа E. Landfeldt и соавт., возраст потери амбулаторности варьирует от 8,6 до 10,3 года [20, 27]. В связи с этими данными можно сделать вывод о положительном эффекте аталурена в виде улучшения или стабилизации двигательной функции пациентов с нмМДД. Стоит отметить, что пациент, потерявший свою амбулаторность, в 17 лет продолжает получать терапию аталуреном в стандартной дозе 40 мг/кг в сутки с последующей коррекцией в зависимости от веса.
Добавим, что при оценке безопасности использования аталурена у всех пациентов отсутствовали серьёзные нежелательные явления, и препарат принимался без перерывов в течение всего периода наблюдения.
Важно также отметить, что эффективный контроль симптомов и замедление прогрессирования заболевания имеют долгосрочные преимущества. Это может привести к меньшему числу госпитализаций, снижению потребности в респираторной поддержке и более высокому уровню участия в социальных и образовательных аспектах жизни.
Заключение
Своевременная диагностика и лечение пациентов в соответствии с клиническими рекомендациями играют ключевую роль в успешной терапии пациентов с МДД. Аталурен не только замедляет ухудшение клинической симптоматики, но также улучшает выживаемость пациентов. Эти результаты подчёркивают важность данного препарата не только в контексте лечения самих больных, но и в отношении их семей, поскольку сохранение независимости в передвижении и дыхательной функции способствует повышению качества жизни и автономии пациентов.
1. Emery A.E. The muscular dystrophies. Lancet. 2002; 359(9307): 687–95. https://doi.org/10.1016/s0140-6736(02)07815-7
2. Jones H., De Vivo D.C., Darras B.T. Neuromuscular Disorders of Infancy, Childhood and Adolescence. A Clinician’s Approach. Oxford: Butterworth-Heinemann; 2003
3. Viggiano E., Ergoli M., Picillo E., Politano L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. HumGenet. 2016;135(7): 685–98. https://doi.org/10.1007/s00439-016-1666-6
4. Crisafulli S., Sultana J., Fontana A., Salvo F., Messina S., Trifirò G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020; 15(1): 141. https://doi.org/10.1186/s13023-020-01430-8
5. Ferlini A., Neri M., Gualandi F. The medical genetics of dystrophinopathies: Molecular genetic diagnosis and its impact on clinical practice. Neuromuscul. Disord. 2013; 23(1): 4–14. https://doi.org/10.1016/j.nmd.2012.09.002
6. Blake D.J., Weir A., Newey S.E., Davies K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002; 82(2): 291–329. https://doi.org/10.1152/physrev.00028.2001
7. Gao Q.Q., McNally E.M. The dystrophin complex: structure, function, and implications for therapy. Compr. Physiol. 2015; 5(3): 1223–39. https://doi.org/10.1002/cphy.c140048
8. Birnkrant D.J., Bushby K., Bann C.M., Apkon S.D., Blackwell A., Brumbaugh D., et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018; 17(3): 251–67. https://doi.org/10.1016/s1474-4422(18)30024-3
9. Клинические рекомендации. Прогрессирующая мышечная дистрофия Беккера; 2023.
10. Marden J.R., Freimark J., Yao Z., Signorovitch J., Tian C., Wong B.L. Real-world outcomes of long-term prednisone and deflazacort use in patients with Duchenne muscular dystrophy: Experience at a single, large care center. J. Comp. Eff. Res. 2020; 9(3): 177–89. https://doi.org/10.2217/cer-2019-0170
11. Angelini C., Peterle E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. 2012; 31(1): 9–15.
12. McDonald C.M., Henricson E.K., Abresch R.T., Duong T., Joyce N.C., Hu F., et al. CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018; 391(10119): 451–61. https://doi.org/10.1016/s0140-6736(17)32160-8
13. Hammer S., Toussaint M., Vollsæter M., Nesbjørg Tvedt M., Drange Røksund O., Reychler G., et al. Exercise training in Duchenne muscular dystrophy: A systematic review and meta-analysis. J. Rehabil. Med. 2022; 54: jrm00250. https://doi.org/10.2340/jrm.v53.985
14. Case L.E., Apkon S.D., Eagle M., Gulyas A., Juel L., Matthews D., et al. Rehabilitation management of the patient with Duchenne muscular dystrophy. Pediatrics. 2018; 142(Suppl. 2): S17–33. https://doi.org/10.1542/peds.2018-0333d
15. Verhaart I.E.C., Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019; 15(7): 373–86. https://doi.org/10.1038/s41582-019-0203-3
16. Государственный реестр лекарственных средств. Available at: https://grls.rosminzdrav.ru/
17. Michorowska S. Ataluren-Promising Therapeutic Premature Termination Codon Readthrough Frontrunner. Pharmaceuticals (Basel). 2021; 14(8): 785. https://doi.org/10.3390/ph14080785
18. Bushby K., Finkel R., Wong B., Barohn R., Campbell C., Comi G.P., et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014; 50(4): 477–87. https://doi.org/10.1002/mus.24332
19. Mercuri E., Osorio A.N., Muntoni F., Buccella F., Desguerre I., Kirschner J., et al. Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne natural history study (2015–2022): 2022 interim analysis. J. Neurol. 2023; 270(8): 3896–913. https://doi.org/10.1007/s00415-023-11687-1
20. Ricotti V., Ridout D.A., Pane M., Main M., Mayhew A., Mercuri E., et al. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J. Neurol. Neurosurg. Psychiatry. 2016; 87(2): 149–55. https://doi.org/10.1136/jnnp-2014-309405
21. Gissy J.J., Johnson T., Fox D.J., Kumar A., Ciafaloni E., van Essen A.J., et al. Delayed onset of ambulation in boys with Duchenne muscular dystrophy: Potential use as an endpoint in clinical trials. Neuromuscul. Disord. 2017; 27(10): 905–10. https://doi.org/10.1016/j.nmd.2017.06.002
22. Ciafaloni E., Fox D.J., Pandya S., Westfield C.P., Puzhankara S., Romitti P.A., et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J. Pediatr. 2009; 155(3): 380–5. https://doi.org/10.1016/j.jpeds.2009.02.007
23. Matthews E., Brassington R., Kuntzer T., Jichi F., Manzur A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016; 2016(5): CD003725. https://doi.org/10.1002/14651858.cd003725.pub4
24. Guglieri M., Bushby K., McDermott M.P., Hart K.A., Tawil R., Martens W.B., et al. Effect of different corticosteroid dosing regimens on clinical outcomes in boys with Duchenne muscular dystrophy: a randomized clinical trial. JAMA. 2022; 327(15): 1456–68. https://doi.org/10.1001/jama.2022.4315
25. Spurney C., Shimizu R., Morgenroth L.P., Kolski H., Gordish-Dressman H., Clemens P.R. CINRG Investigators. Cooperative International Neuromuscular Research Group Duchenne Natural History Study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle Nerve. 2014; 50(2): 250–6. https://doi.org/10.1002/mus.24163
26. Phillips M.F., Quinlivan R.C., Edwards R.H., Calverley P.M. Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am. J. Respir. Crit. Care Med. 2001; 164(12): 2191–4. https://doi.org/10.1164/ajrccm.164.12.2103052
27. Landfeldt E., Alemán A., Abner S., Zhang R., Werner C., Tomazos I., et al. Predictors of loss of ambulation in Duchenne muscular dystrophy: a systematic review and meta-analysis. J. Neuromuscul. Dis. 2024; 11(3): 579–612. https://doi.org/10.3233/jnd-230220
Врач-невролог, зав. 3-м психоневрологическим отд. ГБУЗ МО «НИКИ детства Минздрава Московской области», 141009, г. Мытищи, Россия
e-mail: nakhusheva@mail.ru
Врач-невролог ГБУЗ МО «НИКИ детства Минздрава Московской области», 141009, г. Мытищи, Россия
e-mail: artem.serov@gmail.com
Канд. мед. наук, врач-невролог, зам. директора по международному сотрудничеству ГБУЗ МО «НИКИ детства Минздрава Московской области», 141009, г. Мытищи, Россия
e-mail: ollapochkin@yandex.ru
Нахушева Ф.И., Серов А.В., Лапочкин О.Л. Опыт терапии пациентов с миодистрофией Дюшенна, вызванной нонсенс-мутацией. Неврологический журнал имени Л.О. Бадаляна. 2025;6(3):133-139. https://doi.org/10.46563/2686-8997-2025-6-3-133-139. EDN: bdbbqm
Nakhusheva F.I., Serov A.V., Lapochkin O.L. The experience of treating patients with Duchenne myodystrophy caused by nonsense mutation. L.O. Badalyan Neurological Journal. 2025;6(3):133-139. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-3-133-139. EDN: bdbbqm

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















