


Содержание
Перейти к:
 Валентина Геннадьевна Каверина,
Лейла Ахатовна Гандаева,
Елена Николаевна Басаргина,
Юлия Игоревна Давыдова,
Александр Алексеевич Пушков,
Кирилл Викторович Савостьянов
Валентина Геннадьевна Каверина,
Лейла Ахатовна Гандаева,
Елена Николаевна Басаргина,
Юлия Игоревна Давыдова,
Александр Алексеевич Пушков,
Кирилл Викторович Савостьянов https://doi.org/10.46563/2686-8997-2025-6-2-85-97
EDN: bqwkst
Перейти к:
Введение. RAS-патии — уникальная группа заболеваний с мультисистемным характером поражения. В структуре RAS-патий каузальные генетические варианты гена RAF1 являются наиболее частой причиной гипертрофической кардиомиопатии. Среди всех зарегистрированных вариантов гена RAF1 миссенс-вариант c.770C>T, p.S257L составляет примерно 50% случаев заболевания. В связи с гетерогенностью клинических проявлений у пациентов с патогенными вариантами гена RAF1 установление клинико-генетических особенностей остаётся актуальным.
Цель исследования — установить клинические и молекулярно-генетические характеристики российских детей с синдромом Нунан, обусловленным вариантами гена RAF1.
Материалы и методы. Одноцентровое ретроспективно-проспективное когортное исследование выборки из 98 пациентов с RAS-патиями в возрасте от 1 мес до 18 лет.
Результаты. У 19 (19,0%) из 98 пациентов с RAS-патиями выявлены каузальные варианты в гене RAF1. Все генетические варианты, обнаруженные в гене RAF1, локализуются в кластерной части гена в домене CR2. Вариант c.770C>T, p.S257L являлся причиной заболевания в 10,22% случаев диагностированных нами RAS-патий и у 52,6% детей с вариантами гена RAF1. У всех пациентов с вариантами гена RAF1 уже с первых месяцев жизни отмечались характерные экстракардиальные признаки синдрома Нунан. Гипертрофия миокарда присутствовала у всех пациентов, у подавляющего большинства (89,4%) — с обструкцией выходного тракта левого желудочка. При анализе динамики ремоделирования чаще (78,9%) отмечены случаи прогрессирования гипертрофии. У 57,8% пациентов было зафиксировано одно из неблагоприятных сердечно-сосудистых событий. Для пациентов с вариантом c.770C>T, p.S257L характерно более тяжёлое течение хронической сердечной недостаточности (p = 0,104) с высоким уровнем NTproBNP (p = 0,003) и градиента давления выходного тракта левого желудочка (p = 0,040).
Заключение. Дети с вариантами гена RAF1 демонстрируют классический фенотип синдрома Нунан с наиболее тяжёлым поражением сердечно-сосудистой системы и нуждаются в комплексном обследовании и динамическом с привлечением специалистов различных профилей.
Соблюдение этических стандартов. Исследование одобрено локальным независимым этическим комитетом при ФГАУ «НМИЦ здоровья детей» Минздрава России, выписка из протокола № 4 от 28.04.2022.
Участие авторов:
Каверина В.Г. — написание текста;
Гандаева Л.А. — концепция, редактирование текста;
Басаргина Е.Н. — концепция, редактирование текста;
Давыдова Ю.И. — редактирование текста;
Пушков А.А. — редактирование текста;
Савостьянов К.В. — концепция, редактирование текста.
Все соавторы — утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Конфликт интересов. Авторы статьи подтверждают отсутствия конфликта интересов.
Поступила 03.06.2025
Принята к печати 10.07.2025
Опубликована 20.08.2025
Каверина В.Г., Гандаева Л.А., Басаргина Е.Н., Давыдова Ю.И., Пушков А.А., Савостьянов К.В. Клиническая и молекулярно-генетическая характеристика 19 российских пациентов с синдромом Нунан, обусловленным вариантами в гене RAF1. Неврологический журнал имени Л.О. Бадаляна. 2025;6(2):85-97. https://doi.org/10.46563/2686-8997-2025-6-2-85-97. EDN: bqwkst
Kaverina V.G., Gandaeva L.A., Basargina E.N., Davydova J.I., Pushkov A.A., Savostyanov K.V. Clinical and molecular genetic characteristics of 19 Russian patients with Noonan syndrome caused by variants in the RAF1. L.O. Badalyan Neurological Journal. 2025;6(2):85-97. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-2-85-97. EDN: bqwkst
Введение
RAS-патии — группа врождённых клинически и генетически гетерогенных заболеваний, обусловленных патогенными генетическими вариантами в одном из генов, кодирующих различные компоненты и регуляторы сигнального пути RAS/MAPK, участвующего в процессах дифференцировки, роста, миграции и апоптоза клеток [1].
Одним из наиболее частых представителей RAS-патий является синдром Нунан (СН), распространённость которого в популяции оценивается как 1 : 1000–2500 живых новорождённых. Характерными для синдрома клиническими проявлениями являются черепно-лицевые дизморфии [1, 2], низкий рост, неврологические нарушения, аномалии эктодермы, мочеполовой системы и опорно-двигательного аппарата [3, 4], нарушения свертывания крови и высокая предрасположенность к онкологическим заболеваниям [1, 3, 5], а также широкий спектр патологии сердечно-сосудистой системы.
Около 50% случаев RAS-патий связаны с вариантами гена белковой тирозинфосфатазы нерецепторного типа 11 (PTPN11), реже причиной заболевания являются варианты генов, кодирующих белки гуанин-нуклеотид-обменных факторов (SOS1 (11–13%) и SOS2), киназы RAF (RAF1 (около 5%) и BRAF (менее 2%)), ГТФазы (KRAS, HRAS и NRAS (менее 2%)) и другие компоненты (CBL, LZTR1, NF1, RIT1, SHOC2) [6]. Каузальные варианты в генах, кодирующих нижестоящие киназы пути RAS/MAP, такие как RAF или MEK, как правило, демонстрируют более тяжёлый фенотип [1, 7–9]. Около 65% пациентов с СН, обусловленным каузальными вариантами гена RAF1, формируют гипертрофическую кардиомиопатию (ГКМП), в то время как общая частота ГКМП при СН составляет всего 20% [7–10]. По данным литературы, среди зарегистрированных вариантов гена RAF1 примерно 50% случаев заболевания составляет миссенс-вариант c.770C>T, p.S257L [9, 11, 12], обусловливающий наиболее тяжёлое течение ГКМП с высокой летальностью в раннем возрасте [5, 9, 11].
В настоящее время продолжается работа по выявлению новых молекулярно-генетических причин и определению их эффектов с установлением возможных клинико-генетических характеристик [7, 9, 11–14]. В исследованиях по изучению RAS-патий, связанных с вариантами гена RAF1, проводилась ретроспективная оценка среди пациентов с вариантом c.770C>T, p.S257L среди японской когорты из 18 пациентов [11], китайской когорты из 11 пациентов [12], итальянской когорты из 20 пациентов [9], однако углублённая оценка ГКМП изучена недостаточно, а основное внимание уделено общим фенотипическим проявлениям. В статье мы представляем результаты нашего одноцентрового ретроспективно-проспективного исследования когорты из 19 российских детей с СН, обусловленным вариантами в гене RAF1.
Цель исследования — установить клинические и молекулярно-генетические характеристики российских детей с СН, обусловленным вариантами гена RAF1.
Материалы и методы
Дизайн исследования. Одноцентровое когортное ретроспективно-проспективное исследование выборки из 98 российских детей с генетически подтверждённым синдромом из группы RAS-патий.
Критерии соответствия. Критерии включения: возраст от 1 мес до 18 лет; наличие патологии сердечно-сосудистой системы (кардиомиопатия, врождённые пороки сердца и/или нарушения ритма сердца и проводимости) в сочетании с экстракардиальными особенностями фенотипа (лицевой дизморфизм, низкий рост, короткая шея, крыловидные складки, деформация грудной клетки, крипторхизм, пятна цвета «кофе с молоком» и т. д.); наличие потенциально патогенного генетического варианта (патогенный, вероятно патогенный, вариант неизвестного клинического значения) в одном из генов сигнального RAS/MAPK-пути.
Критерии невключения: отказ от участия в данном исследовании.
Критерии исключения: отказ/невозможность проведения молекулярно-генетического исследования; отсутствие признаков патологии сердечно-сосудистой системы при углублённом лабораторно-инструментальном обследовании; наличие вероятно доброкачественного или доброкачественного варианта в генах RAS/MAPK-пути.
Условия проведения и продолжительность исследования. Исследование проведено на базе кардиологического отделения и лаборатории медицинской геномики Медико-генетического центра ФГАУ «НМИЦ здоровья детей» Минздрава России с 2012 по 2024 г. В связи с тем что часть пациентов на момент включения в исследование уже находилась под наблюдением в кардиологическом отделении ФГАУ «НМИЦ здоровья детей» Минздрава России, у них проводился ретроспективный анализ предшествующих историй болезни в период с 2012 по 2022 г., с 2022 по 2024 г. исследование носило проспективный характер.
Этическая экспертиза. Дизайн исследования был одобрен Этическим комитетом ФГАУ «НМИЦ здоровья детей» Минздрава России № 4 от 28.04.2022.
Назначения и порядок измерений. Всем детям выполнено комплексное обследование, включавшее:
Верификация кардиологических фенотипов проводилась в соответствии с действующими клиническими рекомендациями, клинического синдрома из группы RAS-патий — при наличии нескольких классических фенотипических признаков, включавших: постнатальный низкий рост (SDS рост/возраст < 2 Z-score), гипертелоризм и/или антимонголоидный разрез глаз, макроцефалию, высокий лоб, выступающие лобные бугры, высокий рост волос на лбу, низкопосаженные, ротированные назад ушные раковины, короткую шею/шейные складки, деформацию грудной клетки, задержку моторного развития и/или психоречевого развития, когнитивный дефицит, водянку плода, плевральный выпот, лимфатический отёк, крипторхизм, врождённые аномалии почек, гиперкератоз, надбровную ульэритему, гиперпигментацию кожи, пятна цвета «кофе с молоком», миеломоноцитарные невусы, птоз/полуптоз, тугоухость.
Молекулярно-генетическое исследование в 37 случаях проведено методом высокопроизводительного секвенирования с применением панели генов, разработанной в лаборатории медицинской геномики Медико-генетического центра ФГАУ «НМИЦ здоровья детей» Минздрава России [15], в 66 случаях — путём секвенирования клинического экзома, в 9 — методом секвенирования по Сэнгеру путём исследования кодирующих и прилегающих интронных областей генов PTPN11 и HRAS. Биоинформатический анализ осуществлялся в соответствии рекомендациям GATK Best Practices (https://gatk.broadinstitute.org). Патогенность вариантов, не описанных ранее, определяли при помощи программы «Alamut Visual» («Interactive Biosoftware») со встроенными программными модулями SIFT, PolyPhen HDIV, PolyPhen HVAR, Mutation Taster, FATHMM, CADD13, DANN, M-CAP, REVEL, а также интерпретировали с помощью руководства по интерпретации данных последовательности нуклеотидов ДНК человека [16]. Оценку клинической значимости описанных генетических вариантов давали на основании описания в базе данных HGMD Professional [17].
Статистический анализ. Размер выборки предварительно не рассчитывался. Путём сплошной выборки по результатам клинико-инструментального и молекулярно-генетического исследования нами была отобрано 98 пациентов с патологией сердечно-сосудистой системы и патогенными, вероятно патогенными вариантами и вариантами неизвестного клинического значения в генах пути RAS/MAPK. Отсутствующие данные обрабатывали с помощью метода Complete Case Analysis, при котором строки, содержащие пропуски, исключали из набора данных.
В данной статье для статистического анализа подгруппы из 19 пациентов с вариантами в гене RAF1 нами было проведено разделение их на 2 подгруппы: 1-ю подгруппу составили пациенты с СН, с вариантом c.770C>T, p.S257L, 2-ю подгруппу — пациенты с СН, с другими вариантами гена RAF1.
Статистический анализ проводили с использованием программы «StatTech v. 4.1.2» («Статтех»). Количественные показатели оценивали на предмет соответствия нормальному распределению с помощью критерия Шапиро–Уилка (при числе исследуемых менее 50). В случае описания количественных показателей, имеющих нормальное распределение, полученные данные объединялись в вариационные ряды, в которых проводился расчёт средних арифметических величин (M) и стандартных отклонений (SD), границ 95% доверительного интервала (ДИ). В случае отсутствия нормального распределения количественные данные описывали с помощью медианы (Me) и нижнего и верхнего квартилей [Q1; Q3]. Сравнение 3 и более групп по количественному показателю, распределение которого отличалось от нормального, выполняли с помощью критерия Краскела–Уоллиса, апостериорные сравнения — с помощью критерия Данна с поправкой Холма. Категориальные данные описывали с указанием абсолютных значений и процентных долей. Сравнение процентных долей при анализе многопольных таблиц сопряжённости выполняли с помощью критерия χ2 Пирсона. Различия считали статистически значимыми при p < 0,05. Все результаты приведены с двусторонним уровнем значимости.
Результаты
У 98 пациентов с патологией сердечно-сосудистой системы в 12 генах (BRAF, CBL, HRAS, KRAS, LZTR1, NF1, PTPN11, RAF1, RIT1, SHOC2, SOS1, SOS2), патогенные варианты в которых ранее были описаны в качестве причины RAS-патий, был выявлен 61 патогенный, вероятно патогенный варианты и вариант с неизвестной патогенностью. У 19 (19%) пациентов, из которых 9 (47,4%) мальчики и 10 (52,6%) — девочки, верифицированы патогенные и вероятно патогенные варианты в гене RAF1.
Молекулярно-генетическая характеристика
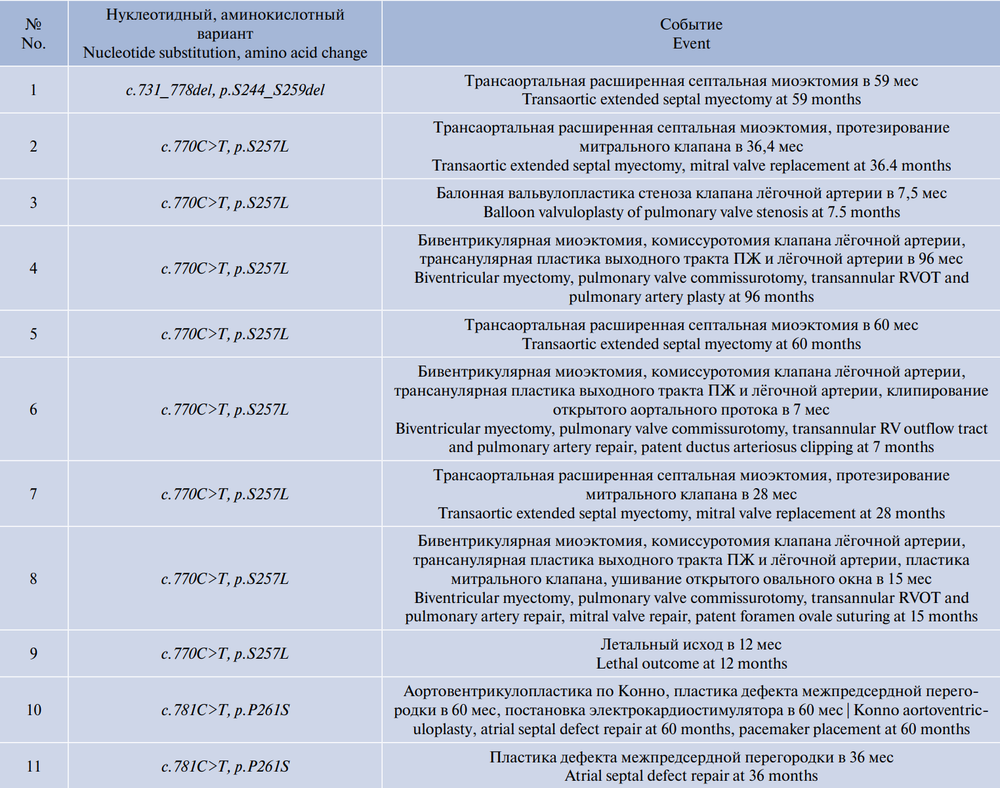
В гене RAF1 было обнаружено 5 патогенных и 1 вероятно патогенный генетический вариант у 19 неродственных пациентов. Выявленные генетические варианты, их локализация и описание в базах данных представлены в табл. 1.
Семейная сегрегация нуклеотидного варианта, обнаруженного у пробанда, методом секвенирования по Сэнгеру в ДНК обоих родителей проведена в 9 (47,3%) из 19 семей. Генетические варианты 731_778del и c.741_776del не были описаны при проведении популяционных исследований. При ранее неописанном варианте c.731_778del (p.S244_S259del) и варианте c.786T>G, p.N262K, отсутствующем в базе HGMD, установлен характер de novo. Семейный характер заболевания выявлен у пациента с вариантом c.781C>T, p.P261S, что составило 11,0% от числа обследованных семей. В остальных 89,0% случаях подтверждён характер de novo. В случае варианта c.741_776del (p.E248_S259del) молекулярно-генетическое исследование проведено только матери пробанда (не выявлено), отец оказался не доступен для анализа. У одного из пациентов с вариантом c.770C>T, p.S257L анамнез был отягощён по ГКМП по линии отца, который также демонстрировал фенотип СН (бивентрикулярная гипертрофия миокарда, птоз, пятна цвета «кофе с молоком»), однако семейный сегрегационный анализ проведён не был в связи с внезапной смертью отца по причине несчастного случая.
Вариант c.770C>T, p.S257L являлся причиной заболевания у 10 пациентов, что составило 10,22% случаев от всех диагностированных нами RAS-патий и 52,6% случаев всех детей с вариантами гена RAF1.
Таблица 1. Спектр и частоты выявленных нуклеотидных вариантов гена RAF1 (NM_002880.4)
Table 1. Spectrum and frequencies of identified nucleotide variants of the RAF1 gene (NM_002880.4)
Клинические характеристики и генотип-фенотипические взаимосвязи
При анализе данных анамнеза установлено, что медиана срока гестации приходится на 39 (37–40) нед. Медиана длины тела при рождении — 51 (49–53) см, массы тела при рождении — 3433,5 (3032–3835) г. У 10 (52,6%) пациентов присутствовали различные сочетания антенатальной патологии (табл. 2).
Медиана возраста на момент первого обращения в ФГАУ «НМИЦ здоровья детей» Минздрава России составила 10,0 [4,0; 12,0] мес, медиана возраста диагностики синдрома из группы RAS-патий — 12,0 [9,0; 12,00]. Медиана срока наблюдения статистически не различалась между группами (р = 0,964), составив 32,5 [12,0; 58,75] мес у пациентов с вариантом c.770C>T, p.S257L и 30,5 [10,5; 59,2] мес у пациентов с другими вариантами.
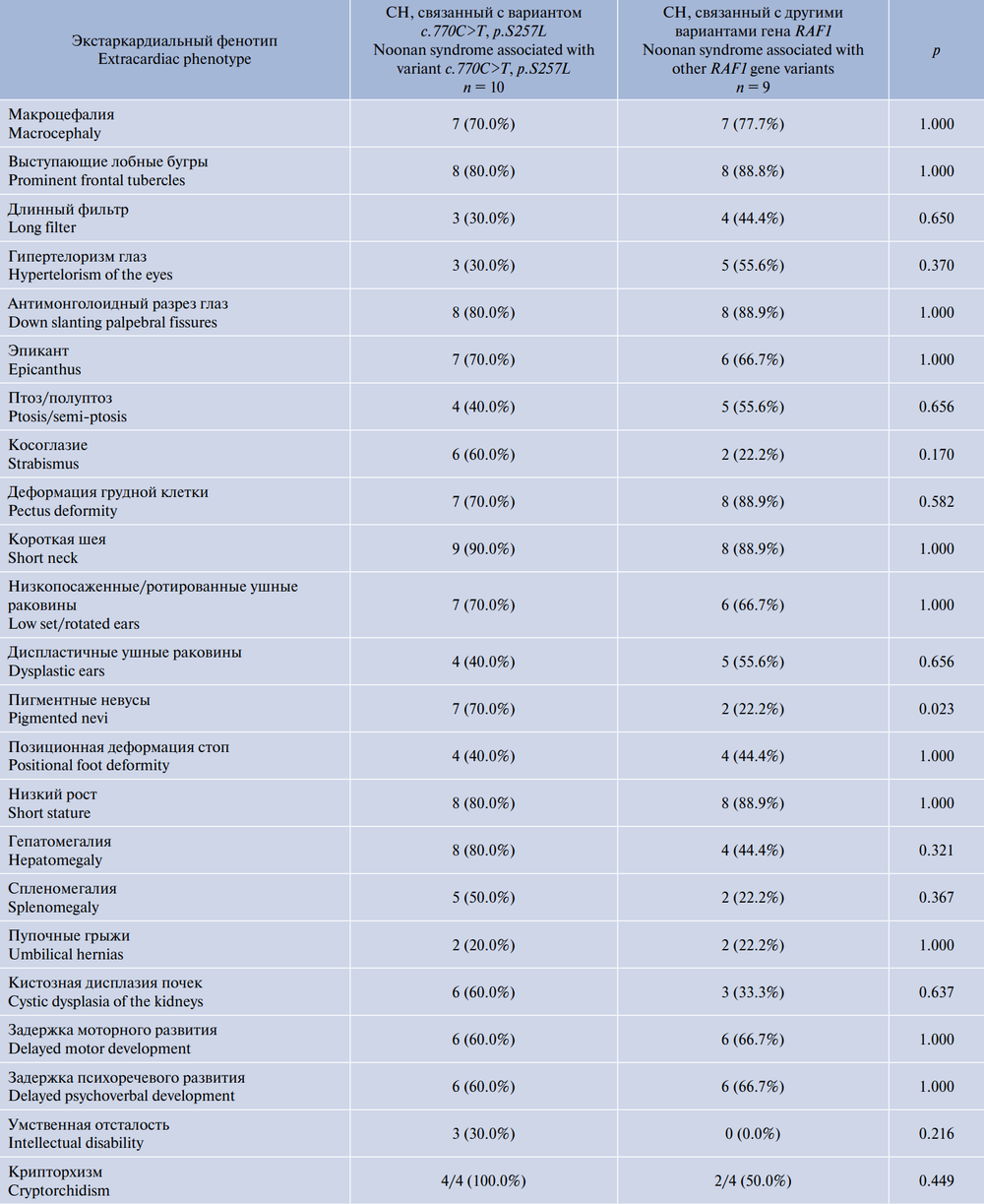
У пациентов с вариантами гена RAF1 уже с первых месяцев жизни отмечались выраженные особенности фенотипа (табл. 3). При первичном обследовании наиболее частыми черепно-лицевыми дисморфиями были макроцефалия, высокий лоб, выступающие лобные бугры, гипертелоризм и антимонголоидный разрез глаз, птоз/полуптоз, эпикант, длинный фильтр, низко посаженные, ротированные и диспластичнные ушные раковины. У всех пациентов отмечена задержка физического развития за счёт низкого роста, у 12 (63,1%) — задержка моторного развития и задержка психоречевого развития, в дальнейшем у 3 (15,7%) из них была диагностирована лёгкая умственная отсталость. У 6 (31,5%) пациентов были отмечены признаки белково-энергетической недостаточности и трудности при кормлении, что в 2 (10,5%) случаях потребовало зондового кормления в 1-й год жизни. Данные признаки между группами не достигли значимых различий. Необходимо отметить, что при клиническом осмотре только у пациентов с вариантом c.770C>T, p.S257L (50,0%; 5 пациентов) отмечена тугоподвижность крупных дистальных суставов конечностей за счёт укорочения сухожилий, что достигло значимой разницы (р = 0,033). Частыми сопутствующими аномалиями в обеих группах были увеличение размеров/признаки кистозной дисплазии почек, гепато- или спленомегалия, пупочные грыжи и у пациентов мужского пола — крипторхизм. У 1 пациента с вариантом c.770C>T, p.S257L, помимо неконструктивной гидроцефалии, выявлены мультикистозная сирингомиелия спинного мозга С4–Th1, аномалия Арнольда–Киари 1-го типа.
Таблица 2. Сравнительная характеристика антенатальной патологии у российских детей с СН, обусловленным различными вариантами гена RAF1 (n = 19)
Table 2. Comparative characteristics of antenatal pathology in Russian children with Noonan syndrome caused by different variants of the RAF1 gene (n = 19)
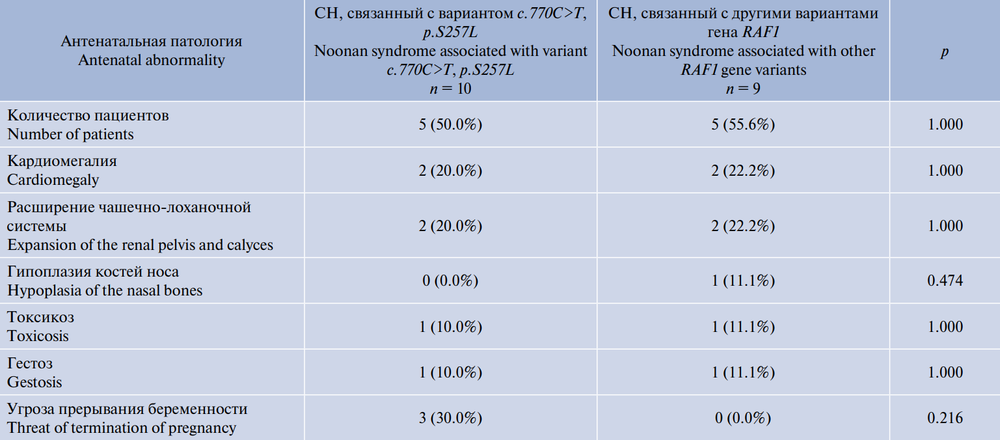
Таблица 3. Сравнительная характеристика основной экстракардиальной патологии у российских детей с СН, обусловленной различными вариантами гена RAF1 (n = 19)
Table 3. Comparative characteristics of the main extracardiac pathology in Russian children with Noonan syndrome caused by different variants of the RAF1 gene (n = 19)
Базовая оценка сердечно-сосудистой системы
На момент первого обращения в ФГАУ «НМИЦ здоровья детей» Минздрава России признаки, соответствующие хронической сердечной недостаточности 2Б или 3 стадии, чаще отмечались у детей с СН, обусловленным вариантом c.770C>T, p.S257L у 8 (80,0%) пациентов, при других вариантах — у 3 (33,3%) (р = 0,104).
Медиана уровня маркера тяжести хронической сердечной недостаточности NTproBNP соответствовала 3376,0 (1512,0–8441,0) пг/мл. Статистически более высокие значения отмечены у пациентов с вариантом c.770C>T, p.S257L: 8441,0 (3688,0–15815,7) против 1465,0 (500,0–3106,0); р = 0,003.
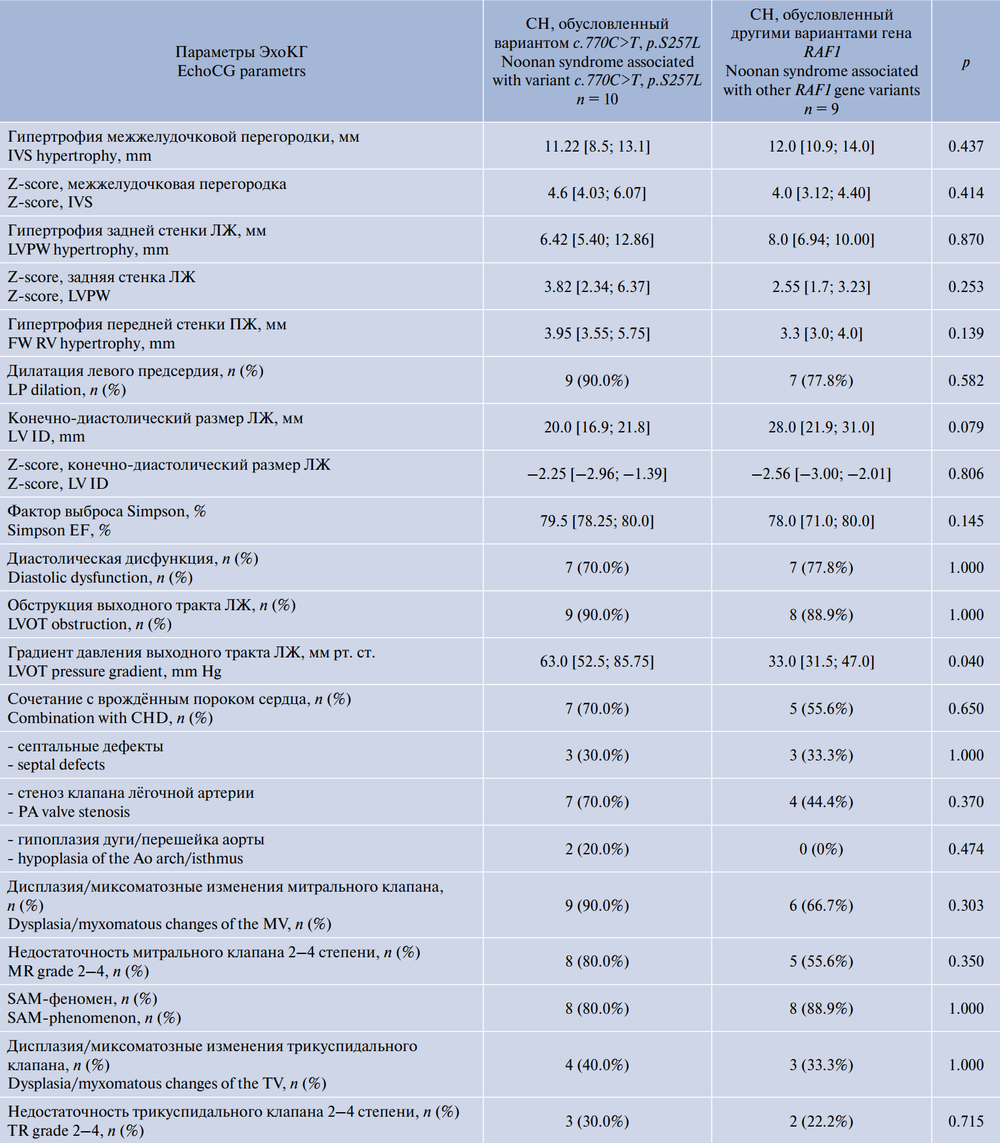
При оценке параметров ЭхоКГ у всех пациентов был диагностирован гипертрофический тип ремоделирования миокарда, в 12 (63,1%) случаях гипертрофия миокарда в комбинации с врождённым пороком сердца. Наиболее часто гипертрофия миокарда сочеталась со стенозом и дисплазией клапана лёгочной артерии (11 (57,8%) пациентов). Также часто встречались дисплазия/миксоматозные изменения клапанного аппарата митрального клапана (15, 78,9%), трикуспидального клапана — у 7 (36,8%), клапана аорты — у 2 (10,5%).
У большинства (n = 12; 63,1%) пациентов гипертрофия миокарда левого желудочка (ЛЖ) имела симметричный характер. Было характерно частое вовлечение в процесс правого желудочка (ПЖ), гипертрофия которого определена у 7 (36,8%) детей. Статистически значимых различий между подгруппами выделить не удалось.
Обструкция выходного тракта ЛЖ отмечена у подавляющего большинства пациентов (n = 17; 89,4%), из них у 4 (21,0%) детей зафиксированы признаки обструкции выходных трактов обоих желудочков, у 8 детей, помимо обструкции выходного тракта ЛЖ, выявлена обструкция и в медиальном сегменте ЛЖ. Степень обструкции статистически была более высокой у больных с вариантом c.770C>T, p.S257L (табл. 4). При сравнении выраженности гипертрофии миокарда у пациентов с вариантом c.770C>T, p.S257L с другими генетическими вариантами значимой разницы не получено.
Таблица 4. Сравнительная характеристика параметров ЭхоКГ у российских детей с различными вариантами гена RAF1, Ме [Q1; Q3] (n = 19)
Table 4. Comparative characteristics of echocardiography parameters in Russian children with different variants of the RAF1 gene, Me [Q1; Q3] (n = 19)
Исходы
Медиана срока наблюдения статистически не различалась между группами (р = 0,964), составив 32,5 [12,0; 58,75] мес у пациентов с вариантом c.770C>T, p.S257L и 30,5 [10,5; 59,2] — у пациентов с другими генетическими вариантами.
За период наблюдения увеличение выраженности симптомов хронической сердечной недостаточности отмечено у 10 пациентов. При анализе динамики ремоделирования гипертрофического фенотипа чаще отмечены случаи прогрессирования (15 (78,9%) пациентов), реже — стабилизации (21,1%).
У 11 (57,8%) пациентов было зафиксировано одно из неблагоприятных сердечно-сосудистых событий, к которым были отнесены летальный исход и необходимость проведения оперативного лечения (табл. 5). Септальная миоэктомия потребовалась 8/19 (50,0%) пациентам, преимущественно (6/8) — с вариантом c.770C>T, p.S257L. В 2 случаях дополнительно проведено протезирование митрального клапана. В раннем послеоперационном периоде 1 пациенту в связи с полной атриовентрикулярной блокадой имплантирован постоянный электрокардиостимулятор. Значительный плевральный/перикардиальный выпот был отмечен у 3 пациентов. При сопоставлении спектра хирургического лечения и генетической причины не удалось установить статистически значимых различий (р = 0,175). Зафиксирован 1 летальный исход: ребёнок с гетерозиготным нуклеотидным вариантом c.770C>T, p.S257L в гене RAF1 умер в возрасте 1 год по причине застойной сердечной недостаточности до получения хирургического лечения.
Таким образом, у пациентов с вариантами гена RAF1 бессобытийная выживаемость через 24 мес составила 57,7 (95% ДИ 29,8–79,1). Различия бессобытийной выживаемости между двумя группами, оценённые с помощью теста отношения правдоподобия, не были значимыми (p = 0,201).
Таблица 5. Неблагоприятные события, отмеченные среди российских детей с вариантами гена RAF1, за время наблюдения
Table 5. Adverse events observed among Russian children with RAF1 gene variants during the observation period
Обсуждение
В российской научной литературе в настоящее время отсутствует информация о частотах различных синдромов RAS-патий, спектре характерных для российской популяции генетических вариантах и информация о взаимосвязи генотипа и фенотипа. В данной статье мы провели сравнение клинических и генетических характеристик у пациентов с СН, связанного с вариантами гена RAF1, ставшего причиной 19,0% случаев всех RAS-патий, наблюдаемых в ФГАУ «НМИЦ здоровья детей» Минздрава России. Данный результат несколько превышает зарубежные данные (5,0–8,0%) [6, 7]. Это отличие может быть связано с включением в нашу выборку только пациентов с патологией сердечно-сосудистой системы, преимущественно с кардиомиопатиями, и, в свою очередь, подтверждает существенный вклад в формирование ГКМП вариантов гена RAF1.
Ген RAF1 кодирует протоонкогенную серин/треонин-протеинкиназу, которая функционирует как эффектор сигнального пути RAS/MAPK [20]. Белок RAF1 состоит из 3 функциональных доменов, называемых консервативными областями 1, 2 и 3 (CR1, CR2 и CR3). Большинство патогенных вариантов RAF1 сгруппированы в домене CR2 экзона 7, состоящего из остатков 251–266, который несёт мотив связывания с 14-3-3 [9, 11, 20, 21]. Две другие кластерные области гена (по 15%) затрагивают остатки в сегменте активации домена киназы CR3 (D486 и T491) или два соседних аминокислотных остатка (S612 и L613), расположенных на С-конце белка. Мутации, затрагивающие мотив связывания 14-3-3 или С-конец белка, приводят к уменьшению фосфорилирования Ser259 и, как следствие, способствуют гиперактивации RAF1 и частичной активации нижестоящих киназ ERK [11, 20, 22]. Мутации, которые группируются в сегменте активации, действуют как доминантно-негативные аллели [11, 20].
В нашем исследовании все генетические варианты, обнаруженные в гене RAF1, локализуются в кластерной части гена в домене CR2. Неописанные в базе данных HGMD варианты c.731_778del, p.S244_S259del и c.741_776del, p.E248_S259del также расположены в области критической для фосфорилирования Ser259 и отсутствуют у обследованных фенотип-негативных родителей пробандов, что позволяет нам расценить их как причину заболевания.
Преимущественно варианты гена RAF1, приводящие к гиперактивации киназы, тесно связаны (84,2–95,0%) с ГКМП [9, 11, 21, 23], что согласуется с результатами нашей работы.
При оценке клинических проявлений у детей, геном которых содержит варианты гена RAF1, наличие ГКМП сочеталось с наиболее тяжёлым течением с выраженными проявлениями гемодинамических нарушений. Также характерными особенностями являлись значительное уменьшение полости ЛЖ, дилатация левого предсердия, диастолическая дисфункция и аномалии строения клапанного аппарата митрального клапана, что рассматривается как фактор риска осложнений ГКМП.
Известно, что более 50% случаев СН, обусловленных патогенными вариантами гена RAF1, затрагивают аминокислотный остаток [9–11, 24]. В нашей работе также превалирует миссенс-вариант c.770C>T, p.S257L, обусловивший заболевание у 10 (52,6% от пациентов с вариантом гена RAF1 и 10,2% от общего числа пациентов) детей. Такой высокий процент может свидетельствовать о том, что данный вариант является мажорным и для российской популяции.
Ряд сообщений свидетельствуют о том, что у пациентов с вариантом c.770C>T, p.S257L обычно развивается особенно тяжёлая и быстро прогрессирующая ГКМП, подразумевающая чрезвычайно высокую и раннюю смертность [9, 11, 12]. Наши результаты подтверждают, что данный генетический вариант связан с наиболее тяжёлым сердечным фенотипом.
Важным маркером оценки тяжести состояния пациентов с ГКМП является наличие обструкции выходного тракта ЛЖ [7, 8, 25, 26]. В нашей работе отмечена высокая распространённость обструктивных форм как при варианте c.770C>T, p.S257L, так и при других вариантах гена RAF1. Высокий градиент обструкции выходного тракта ЛЖ, частая гемодинамически значимая недостаточность митрального клапана за счёт аномалии строения клапанного аппарата и SAM-феномен (систолическое движение передней митральной створки), атриомегалия и клинические признаки хронической сердечной недостаточности регистрировались у большинства пациентов уже с раннего возраста.
Ведение пациентов с кардиоваскулярной патологией при RAS-патиях на сегодняшний день основано на текущих клинических рекомендациях и аналогично методам, используемым в целом в популяции [8, 13, 25, 26]. В случае развития ГКМП в качестве препаратов 1-й линии рекомендуются β-блокаторы с использованием неселективных форм [8, 13]. Трансаортальная расширенная септальная миоэктомия является наиболее предпочтительным вариантом хирургического лечения при обструктивной форме ГКМП с градиентом давления более 50 мм рт. ст. в случае резистентности к медикаментозной терапии [8, 25, 26].
В нашей работе половине пациентов с вариантами гена RAF1 уже в раннем возрасте потребовалась септальная миоэктомия, преимущественно при варианте c.770C>T, p.S257L. Полученные положительные результаты подтверждают безопасность и эффективность данной процедуры для пациентов с ГКМП в структуре RAS-патий.
При выявлении гипертрофии миокарда у детей важно учитывать вероятность сочетания с рядом характерных экстракардиальных признаков, требующих исключения RAS-патий [1, 7, 8]. В нашей работе у всех пациентов с вариантами в гене RAF1 присутствовали ярко выраженные фенотипические признаки: макроцефалия, выступающие лобные бугры, гипертелоризм глазных щелей, эпикант, низко посаженные ушные раковины, крыловидная шея, широкая грудная клетка, что согласуется с зарубежными сообщениями [4, 8, 9, 11, 12].
Интересно, что в нашей когорте пациентов имеются признаки, которые можно охарактеризовать как дефект соединительной ткани (множественные грыжи, тугоподвижность дистальных суставов конечностей, миопия, в том числе изменение клапанного аппарата атриовентрикулярных клапанов), также было характерно увеличение линейных размеров почек или кистозная дисплазия почек, гепато- и спленомегалия, не коррелирующие с вовлечением сердечно-сосудистой системы). Подобные наблюдения ранее не приводились.
В литературе описана высокая частота встречаемости низкого роста у пациентов с вариантом c.770C>T, p.S257L [9, 11]. Это может быть связано как с выраженностью хронической сердечной недостаточности, так и с нарушениями гормональной регуляции. В нашей работе низкий рост отмечен у 16 (84,2%) из 18 пациентов, как с вариантом c.770C>T, p.S257L, так и с другими вариантами (р = 0,477). Касаясь тактики ведения пациентов с RAS-патиями, следует упомянуть о попытке лечения задержки роста рекомбинантным гормоном роста человека, т. к. это является спорным вопросом как в отношении эффективности, так и в отношении влияния на сердечную функцию. В литературе описано применение рекомбинантной терапии человеческим гормоном роста у 4 пациентов с вариантом c.770C>T, p.S257L, двое из которых прекратили лечение из-за документально подтверждённого ухудшения ГКМП в виде нарастания толщины миокарда и степени обструкции [9].
Описана предрасположенность пациентов с вариантами гена RAF1 к гиперпигментации кожных покровов [9, 11]. У наших пациентов в 47,3% случаев отмечено наличие миеломоноцитарных невусов, в 10,4% — пятен цвета «кофе с молоком».
В итальянском исследовании среди пациентов с вариантом c.770C>T, p.S257L в большом (40%) проценте случаев зафиксированы аномалии ЦНС [9, 27]. В основном встречались мальформация Арнольда–Киари I типа, обструктивная гидроцефалия или вентрикуломегалия. Эпилепсия и аномалии электроэнцефалографии или расстройства аутистического спектра наблюдались у 6% пациентов.
В нашей работе вентрикуломегалия также присутствовала у значительной доли пациентов (5 детей; 26,3%) как с вариантом c.770C>T, p.S257L, так и с другими вариантами. У 1 пациента с вариантом c.770C>T, p.S257L диагностирована аномалия Арнольда–Киари 1 типа в сочетании с мультикистозной сирингомиелией спинного мозга. Случаи эпилепсии или расстройства аутистического спектра отсутствовали.
Пациенты с вариантами в RAF1 демонстрируют различные когнитивные функции: от лёгкой умственной отсталости (55%) до нормальных интеллектуальных способностей [27]. В нашей работе когнитивные нарушения присутствовали у меньшего числа пациентов (15,7%).
Для антенатальных аномалий пациентов с СН, обусловленных патогенными вариантами гена RAF1, характерно увеличение толщины воротникового пространства или многоводие, особенно у пациентов с вариантом c.770C>T, p.S257L (26 и 47% соответственно) [9], в нашей работе подобные признаки регистрировались в обеих группах.
Среди обследованных нами детей не отмечено случаев формирования злокачественных новообразований, однако возникновение опухолей при вариантах гена RAF1 соответствует общей распространённости при СН (около 2%) [1, 9], что делает необходимым онкологическую настороженность на протяжении всей жизни пациентов.
Значительный процент случаев является результатом мутаций de novo, как и для других редких болезней с доминантным типом наследования. Однако примерно в 30–70% случаев заболевание носит семейный характер [1]. В нашей работе наследственный характер заболевания подтверждён в 1 из 9 обследованных семей, что составляет лишь 11,0% случаев. Это подтверждает необходимость обязательного медико-генетического консультирования пациентов и их семей при подозрении на заболевания группы RAS-патий.
Заключение
В статье мы представляем клинико-генетические характеристики 19 российских детей с СН, обусловленным вариантами в гене RAF1. Наша работа подтверждает важность установления этиологической причины RAS-патий путём проведения молекулярно-генетической диагностики в связи с многообразием клинических признаков и риском семейных форм заболевания.
Особое внимание нами было уделено поражению сердечно-сосудистой системы. Нами подтверждено, что каузальные варианты гена RAF1, связанные с ГКМП, преимущественно локализуются в кластерной части гена в домене CR2. При анализе особенностей ГКМП установлено, что пациенты с вариантом c.770C>T, p.S257L, который является мажорным для российской популяции, уже с раннего возраста демонстрируют более тяжёлый фенотип с выраженными признаками хронической сердечной недостаточности, статистически более высоким уровнем NTproBNP и градиента давления выходного тракта ЛЖ. Более половины этих пациентов нуждаются в раннем оперативном лечении.
Результаты нашего исследования в сочетании с данными литературы указывают на то, что дети с различными вариантами гена RAF1 демонстрируют классический фенотип СН, который затрагивает множество органов и систем и, таким образом, на протяжении всей жизни нуждаются в комплексном обследовании и динамическом наблюдении в условиях многопрофильного лечебного учреждения с привлечением специалистов различных профилей.
1. Rauen K.A. Defining RASopathy. Dis. Model. Mech. 2022; 15(2): dmm049344. https://doi.org/10.1242/dmm.049344
2. Tartaglia M., Gelb B.D., Zenker M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2011; 25(1): 161–79. https://doi.org/10.1016/j.beem.2010.09.002
3. Leoni C., Blandino R., Delogu A.B., De Rosa G., Onesimo R., Verusio V., et al. Genotype-cardiac phenotype correlations in a large single-center cohort of patients affected by RASopathies: Clinical implications and literature review. Am. J. Med. Genet. A. 2022; 188(2): 431–45. https://doi.org/10.1002/ajmg.a.62529
4. Stevenson D.A., Yang F.C. The musculoskeletal phenotype of the RASopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2011; 157(2): 90–103. https://doi.org/10.1002/ajmg.c.30296
5. Romano A.A., Allanson J.E., Dahlgren J., Gelb B.D., Hall B., Pierpont M.E., et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010; 126(4): 746–59. https://doi.org/10.1542/peds.2009-3207
6. Hilal N., Chen Z., Chen M.H., Choudhury S. RASopathies and cardiac manifestations. Front. Cardiovasc. Med. 2023; 10: 1176828. https://doi.org/10.3389/fcvm.2023.1176828
7. Calcagni G., Limongelli G., D’Ambrosio A., Gesualdo F., Digilio M.C., Baban A., et al. Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int. J. Cardiol. 2017; 245: 92–8. https://doi.org/10.1016/j.ijcard.2017.07.068
8. Lioncino M., Monda E., Verrillo F., Moscarella E., Calcagni G., Drago F., et al. Hypertrophic cardiomyopathy in RASopathies: Diagnosis, clinical characteristics, prognostic implications, and management. Heart Fail. Clin. 2022; 18(1): 19–29. https://doi.org/10.1016/j.hfc.2021.07.004
9. Gazzin A., Fornari F., Niceta M., Leoni C., Dentici M.L., Carli D., et al. Defining the variant-phenotype correlation in patients affected by Noonan syndrome with the RAF1:c.770C>T p.(Ser257Leu) variant. Eur. J. Hum. Genet. 2024; 32(8): 964–71. https://doi.org/10.1038/s41431-024-01643-6
10. Nakhaei-Rad S., Haghighi F., Bazgir F., Dahlmann J., Busley A.V., Buchholzer M., et al. Molecular and cellular evidence for the impact of a hypertrophic cardiomyopathy-associated RAF1 variant on the structure and function of contractile machinery in bioartificial cardiac tissues. Commun. Biol. 2023; 6(1): 657. https://doi.org/10.1038/s42003-023-05013-8
11. Kobayashi T., Aoki Y., Niihori T., Cavé H., Verloes A., Okamoto N., et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum. Mutat. 2010; 31(3): 284–94. https://doi.org/10.1002/humu.21187
12. Chen H., Li X., Liu X., Wang J., Zhang Z., Wu J., et al. Clinical and mutation profile of pediatric patients with RASopathy-associated hypertrophic cardiomyopathy: results from a Chinese cohort. Orphanet J. Rare Dis. 2019; 14(1): 29. https://doi.org/10.1186/s13023-019-1010-z
13. Гандаева Л.А., Каверина В.Г., Басаргина Е.Н., Пушков А.А., Савостьянов К.В. Редкий случай синдрома Нунан, обусловленный биаллельными вариантами в гене LZTR1. Неврологический журнал имени Л.О. Бадаляна. 2023; 4(3): 120–9. https://doi.org/10.46563/2686-8997-2023-4-3-120-129 https://elibrary.ru/laemcj
14. Савостьянов К.В. Современные алгоритмы генетической диагностики редких наследственных болезней у российских пациентов: Информационные материалы. М.: Полиграфист и издатель; 2022. https://elibrary.ru/rduzgh
15. Савостьянов К.В., Намазова-Баранова Л.С., Басаргина Е.Н., Вашакмадзе Н.Д., Журкова Н.В., Пушков А.А. и др. Новые варианты генома российских детей с генетически обусловленными кардиомиопатиями, выявленные методом массового параллельного секвенирования. Вестник Российской академии медицинских наук. 2017; 72(4): 242–53. https://doi.org/10.15690/vramn872 https://elibrary.ru/zfourx
16. Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б., Коновалов Ф.А., Масленников А.Б., Степанов В.А. и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2). Медицинская генетика. 2019; 18(2): 3–23. https://doi.org/10.25557/2073-7998.2019.02.3-23 https://elibrary.ru/jzljue
17. Human Gene Database (HGMD). Available at: https://www.hgmd.cf.ac.uk/docs/new_back.html
18. Chen S., Hu J., Xu Y., Yan J., Li S., Chen L., et al. Transcriptome analysis of human hypertrophic cardiomyopathy reveals inhibited cardiac development pathways in children. iScience. 2023; 27(1): 108642. https://doi.org/10.1016/j.isci.2023.108642
19. Rodriguez F., Ponce D., Berward F.J., Lopetegui B., Cassorla F., Aracena M. RAF1 variant in a patient with Noonan syndrome with multiple lentigines and craniosynostosis. Am. J. Med. Genet A. 2019; 179(8): 1598–602. https://doi.org/10.1002/ajmg.a.61203
20. Tartaglia M., Zampino G., Gelb B.D. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol. Syndromol. 2010; 1(1): 2‐26. https://doi.org/10.1159/000276766
21. Pandit B., Sarkozy A., Pennacchio L.A., Carta C., Oishi K., Martinelli S., et al. Gain‐of‐function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007; 39(8): 1007–12. https://doi.org/10.1038/ng2073
22. Aizaki K., Sugai K., Saito Y., Nakagawa E., Sasaki M., Aoki Y., et al. Cardio-facio-cutaneous syndrome with infantile spasms and delayed myelination. Brain Dev. 2011; 33(2): 166–9. https://doi.org/10.1016/j.braindev.2010.03.008
23. Razzaque M.A., Nishizawa T., Komoike Y., Yagi H., Furutani M., Amo R., et al. Germline gain‐of‐function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007; 39(8): 1013–7. https://doi.org/10.1038/ng2078
24. Jaffre F., Miller C.L., Schänzer A., Evans T., Roberts A.E., Hahn A., et al. Inducible pluripotent stem cell-derived cardiomyocytes reveal aberrant extracellular regulated kinase 5 and mitogen-activated protein kinase kinase 1/2 signaling concomitantly promote hypertrophic cardiomyopathy in RAF1-associated Noonan syndrome. Circulation. 2019; 140(3): 207–24. https://doi.org/10.1161/circulationaha.118.037227
25. Wolf C.M., Zenker M., Burkitt-Wright E., Edouard T., García-Miñaúr S., Lebl J., et al. Management of cardiac aspects in children with Noonan syndrome – results from a European clinical practice survey among paediatric cardiologists. Eur. J. Med. Genet. 2022; 65(1): 104372. https://doi.org/10.1016/j.ejmg.2021.104372
26. Kaltenecker E., Schleihauf J., Meierhofer C., Shehu N., Mkrtchyan N., Hager A., et al. Long-term outcomes of childhood onset Noonan compared to sarcomere hypertrophic cardiomyopathy. Cardiovasc. Diagn. Ther. 2019; 9(Suppl. 2): 299–309. https://doi.org/10.21037/cdt.2019.05.01
27. Wingbermühle E., Roelofs R.L., Oomens W., Kramer J., Draaisma J.M.T., Leenders E., et al. Cognitive phenotype and psychopathology in Noonan syndrome spectrum disorders through various Ras/MAPK pathway associated gene variants. J. Clin. Med. 2022; 11(16): 4735. https://doi.org/10.3390/jcm11164735
Мл. науч. сотр., врач-педиатр ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: coverina.v@yandex.ru
Канд. мед. наук, ведущ. науч. сотр., врач детский кардиолог ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: dr.gandaeva@gmail.com
Доктор мед. наук, профессор, гл. науч. сотр., зав. отделением кардиологии ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: basargina@nczd.ru
Мл. науч. сотр., врач-генетик ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: davydova.iui@nczd.ru
Канд. биол. наук, вед. науч. сотр. лаб. медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: pushkovaa@nczd.ru
Доктор биол. наук, рук. Медико-генетического центра и лаборатории медицинской геномики ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
e-mail: 7443333@gmail.com
Каверина В.Г., Гандаева Л.А., Басаргина Е.Н., Давыдова Ю.И., Пушков А.А., Савостьянов К.В. Клиническая и молекулярно-генетическая характеристика 19 российских пациентов с синдромом Нунан, обусловленным вариантами в гене RAF1. Неврологический журнал имени Л.О. Бадаляна. 2025;6(2):85-97. https://doi.org/10.46563/2686-8997-2025-6-2-85-97. EDN: bqwkst
Kaverina V.G., Gandaeva L.A., Basargina E.N., Davydova J.I., Pushkov A.A., Savostyanov K.V. Clinical and molecular genetic characteristics of 19 Russian patients with Noonan syndrome caused by variants in the RAF1. L.O. Badalyan Neurological Journal. 2025;6(2):85-97. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-2-85-97. EDN: bqwkst

119991, Россия, г. Москва, Ломоносовский проспект, д.2, стр.1
e-mail: neuro.journal@mail.ru
tel.: +7 916 885 28 02























