


Содержание
Перейти к:
 Дарья Андреевна Фисенко,
Алексей Львович Куренков,
Людмила Михайловна Кузенкова,
Владислав Владимирович Черников,
Евгения Владимировна Увакина,
София Георгиевна Попович,
Бэлла Ибрагимовна Бурсагова,
Луизат Муслимовна Абдуллаева,
Юлия Александровна Курова,
Надежда Сергеевна Адалимова,
Дарья Сергеевна Николенко,
Оксана Валерьевна Глоба,
Наталья Владимировна Андреенко
Дарья Андреевна Фисенко,
Алексей Львович Куренков,
Людмила Михайловна Кузенкова,
Владислав Владимирович Черников,
Евгения Владимировна Увакина,
София Георгиевна Попович,
Бэлла Ибрагимовна Бурсагова,
Луизат Муслимовна Абдуллаева,
Юлия Александровна Курова,
Надежда Сергеевна Адалимова,
Дарья Сергеевна Николенко,
Оксана Валерьевна Глоба,
Наталья Владимировна Андреенко https://doi.org/10.46563/2686-8997-2025-6-1-13-25
EDN: yyenrs
Перейти к:
Введение. Появление патогенетической терапии значительно изменило прогнозы при спинальной мышечной атрофии (СМА). Однако наши знания о патогенетических методах лечения СМА в значительной степени базируются на результатах клинических исследований, которые во многом ограничены из-за узких критериев отбора пациентов и небольшой продолжительности наблюдения.
Цель исследования — оценить эффективность генной терапии препаратом онасемноген абепарвовек (ОА) у пациентов с СМА I типа с клиническими проявлениями болезни и у детей, находящихся в пресимптоматической стадии заболевания, в рамках реальной клинической практики.
Материалы и методы. В исследование были включены 79 детей со СМА, из них 42 (53,2%) мальчика. Диагноз был верифицирован при проведении молекулярно-генетического исследования. Дети были распределены на две группы в зависимости от наличия или отсутствия у них симптомов СМА на момент включения в исследование: 44 ребёнка имели СМА I типа с дебютом клинических симптомов в возрасте до 6 мес; 35 детей находились на пресимптоматической стадии заболевания. Все пациенты получили ОА, средний возраст на момент проведения генной терапии составил 2,90 ± 1,74 мес (95% ДИ 2,51–3,29), min — 1,00, max — 7,00. Комплексная оценка клинических показателей (этапы двигательного развития по рекомендациям ВОЗ, шкалы HINE-2 и CHOP-INTEND) осуществлялась до инициации генной терапии и через 6, 12, 18 и 24 мес после её проведения.
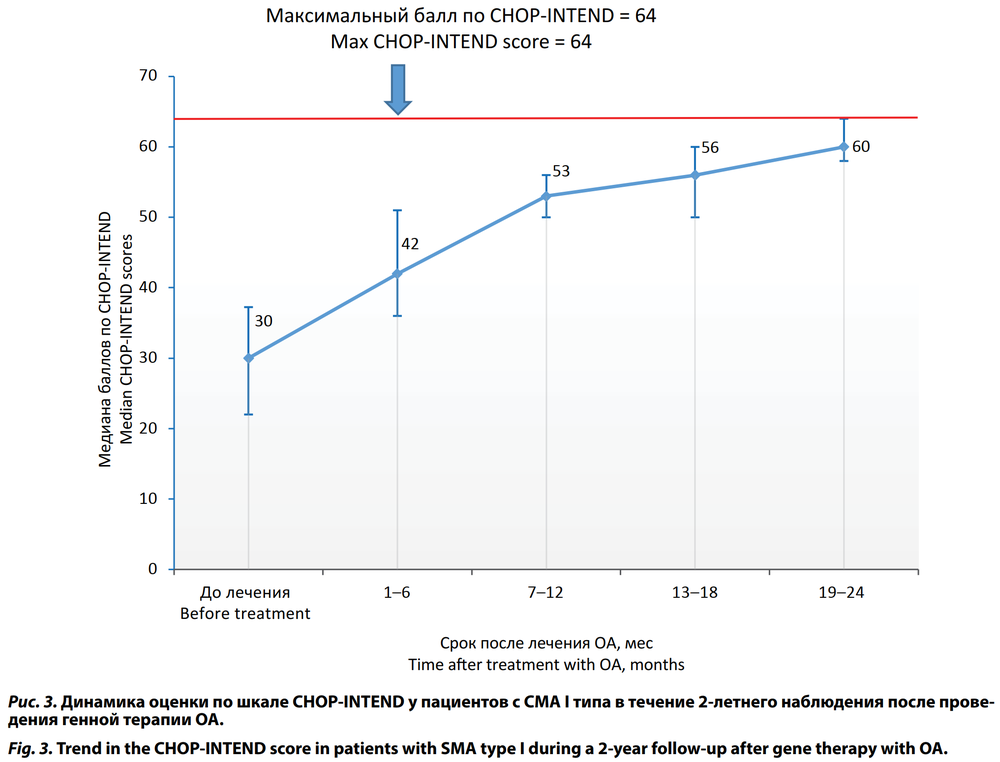
Результаты. Дети с СМА I типа после проведения генной терапии демонстрировали положительную динамику в двигательном развитии. К концу 2-го года наблюдения хорошо удерживали голову и переворачивались из положения лёжа на спине на живот 88,6% пациентов; сидели без опоры — 60,5%, но только 10,3% смогли сформировать все двигательные навыки, однако ни один из этих детей не сформировал их в соответствии с критериями ВОЗ. Оценка по шкале HINE-2 увеличилась с Me 2,0 (1,00–2,25) баллов при инициации терапии до 20,00 (16,50–24,50) баллов к концу периода наблюдения. Максимального значения 26 баллов достигли только 2 (4,5%) детей этой группы. Оценка по шкале CHOP-INTEND возросла с Me 30,0 (22,00–37,25) баллов перед началом лечения до Me 60,0 (58,0–64,0) баллов к 24 мес наблюдения. Только 11,4% пациентов с СМА I типа достигли максимального значения 64 балла.
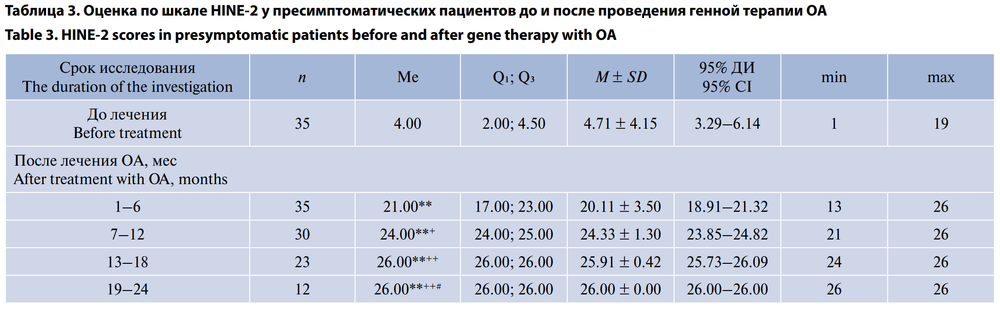
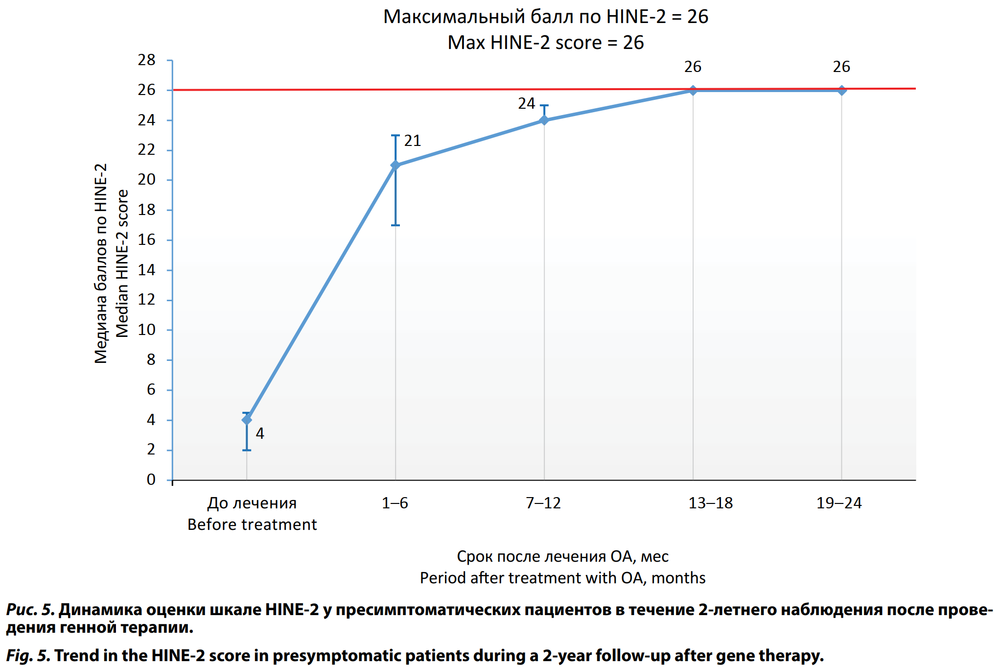
Большинство пресимптоматических пациентов в нашем исследовании на фоне генной терапии достигли всех двигательных навыков по критериям ВОЗ соответственно своему возрасту. Все дети из этой группы, которые находились под достаточным наблюдением и достигли возраста самостоятельного хождения (23 ребёнка), имели максимальное значение 26 баллов по шкале HINE-2 к возрасту 18 мес. Все дети из этой группы достигли максимального значения по шкале CHOP-INTEND 64 балла к возрасту 6 мес.
Заключение. Применение ОА у детей с СМА I типа существенно модифицирует течение заболевания и значимо улучшает исходы по сравнению с естественной историей развития СМА с дебютом в раннем возрасте, а у большинства пресимптоматических пациентов использование генной терапии приводит к достижению этапов двигательного развития в соответствии с критериями ВОЗ.
Соблюдение этических стандартов. На проведение данного исследования было получено разрешение локального этического комитета ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России (протокол заседания № 10 от 06.10.2022).
Участие авторов:
Фисенко Д.А. — концепция и дизайн статьи, написание текста, редактирование;
Куренков А.Л. — концепция и дизайн статьи, написание текста, редактирование;
Кузенкова Л.М. — концепция и дизайн статьи, редактирование;
Черников В.В. — статистическая обработка данных;
Увакина Е.В. — концепция и дизайн статьи, редактирование;
Попович С.Г. — редактирование;
Бурсагова Б.И. — редактирование;
Абдуллаева Л.М. — редактирование;
Курова Ю.А. — редактирование;
Адалимова Н.С. — редактирование;
Николенко Д.С. — редактирование;
Глоба О.В. — редактирование;
Андреенко Н.В. — редактирование.
Все соавторы — утверждение окончательного варианта статьи, ответственность за целостность всех частей статьи.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов в связи с публикацией данной статьи.
Финансирование. Исследование не имело спонсорской поддержки.
Поступила 12.03.2025
Принята к печати 07.04.2025
Опубликована 30.04.2025
Фисенко Д.А., Куренков А.Л., Кузенкова Л.М., Черников В.В., Увакина Е.В., Попович С.Г., Бурсагова Б.И., Абдуллаева Л.М., Курова Ю.А., Адалимова Н.С., Николенко Д.С., Глоба О.В., Андреенко Н.В. Эффективность генной терапии препаратом онасемноген абепарвовек у пациентов со спинальной мышечной атрофией раннего возраста. Неврологический журнал имени Л.О. Бадаляна. 2025;6(1):13-25. https://doi.org/10.46563/2686-8997-2025-6-1-13-25. EDN: yyenrs
Fisenko D.A., Kurenkov A.L., Kuzenkova L.M., Chernikov V.V., Uvakina E.V., Popovich S.G., Bursagova B.I., Abdullaeva L.M., Kurova J.A., Adalimova N.S., Nikolenko D.S., Globa O.V., Andreenko N.V. The efficacy of gene therapy with onasemnogene abeparvovec in spinal muscular atrophy in young patients. L.O. Badalyan Neurological Journal. 2025;6(1):13-25. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-1-13-25. EDN: yyenrs
Введение
Спинальная мышечная атрофия (СМА) — это наследственное нервно-мышечное заболевание, которое характеризуется нейродегенерацией двигательных нейронов, что приводит к прогрессирующей мышечной слабости, атрофии скелетных и бульбарных мышц [1, 2]. Клинические проявления СMA имеют генетическую природу и обусловлены мутациями в гене SMN1. Тяжесть заболевания различна. При этом число копий гена SMN2 имеет сильную корреляцию с началом болезни и тяжестью СМА. Это позволяет рассматривать число копий гена SMN2 как важный фенотипический модификатор заболевания [3]. Исторически фенотип СMA классифицируется по 5 клиническим типам в зависимости от возраста начала заболевания и максимальной двигательной функции — от 0 типа (наиболее тяжёлого, при внутриутробном развитии заболевания) до 4 типа (наименее тяжёлого, при более позднем начале заболевания) [4, 5]. При СМА 1-го типа дебют заболевания отмечается в течение первых 6 мес жизни и наблюдается быстрое прогрессирование болезни. На долю СМА 1-го типа приходится примерно 60% случаев заболевания [2].
СМА до недавнего времени рассматривалась как одна из ведущих генетических причин ранней детской смертности, т. к. при естественном течении заболевания при СМА 1-го типа почти все пациенты (90%) умирают или нуждаются в постоянной вентиляции лёгких к 2 годам [6]. С появлением патогенетической терапии СМА прогнозы значительно улучшились [7–9]. В 2019 г. Управление по контролю за продуктами и лекарственными средствами США одобрило к применению онасемноген абепарвовек (ОА) — одноразовое внутривенное введение векторной генной терапии на основе аденоассоциированного вируса 9 (AAV9), что позволило доставлять полностью функциональную копию комплементарной ДНК гена SMN1 человека в клетки-мишени [10, 11].
Знания об этих новых патогенетических методах лечения СМА в значительной степени базируются на результатах клинических исследований, которые ограничены, в первую очередь, узкими критериями отбора и ограниченной продолжительностью наблюдения.
В 2023 г. в России стартовал неонатальный скрининг, который способствовал раннему выявлению пациентов с СМА, что привело к возможности более широкого применения генной терапии в реальной клинической практике.
Цель исследования — оценить эффективность генной терапии препаратом ОА у пациентов с СМА I типа с клиническими проявлениями болезни и у детей, находящихся в пресимптоматической стадии заболевания.
Материалы и методы
В исследование были включены 79 детей с СМА, из них 42 (53,2%) мальчика. Диагноз был верифицирован при проведении молекулярно-генетического исследования — у всех детей была выявлена делеция экзонов 7 и/или 8 гена SMN1 в гомозиготном состоянии. Все пациенты получили генную терапию препаратом ОА, средний возраст на момент проведения терапии составил 2,90 ± 1,74 мес (95% ДИ 2,51–3,29), min — 1,00, max — 7,00.
Все дети на момент рождения имели гестационный возраст 37–40 нед, не имели отягощённого перинатального анамнеза (оценка по шкале Aпгар при рождении — 8 баллов и выше; вес — 2400 г и более; дети не имели признаков асфиксии и гипоксии; не требовали перевода в отделение реанимации и интенсивной терапии; не имели видимых пороков развития, не требовали экстренного или планового хирургического лечения в раннем возрасте).
До включения в исследование детей осматривал педиатр для исключения заболеваний желудочно-кишечного тракта, дыхательной, сердечно-сосудистой и мочеполовой систем, не связанных с течением основного заболевания. Для оценки двигательного, психического, предречевого и речевого развития всех пациентов осматривал невролог по стандартной методике [12].
Дети были распределены на две группы в зависимости от наличия или отсутствия у них симптомов СМА на момент включения в исследование. В 1-ю группу были отнесены 44 ребёнка (22 мальчика и 22 девочки) с клиническими симптомами заболевания, манифестировавшего у них в возрасте до 6 мес жизни, что обусловило целесообразность постановки всем детям диагноза СМА I типа. У 22 (50%) пациентов диагноз СМА I типа был верифицирован в связи с развитием клинической картины заболевания до внедрения расширенного неонатального скрининга (до 01.01.2023), у 22 (50%) детей диагноз СМА был установлен в рамках расширенного неонатального скрининга, и клинические проявления заболевания манифестировали в первые месяцы жизни.
Вторую группу составили 35 детей (20 (57,1%) мальчиков и 15 (42,9%) девочек) с генетически верифицированным диагнозом СМА без развития симптомов заболевания. На момент включения в исследование и проведения лечения с применением генной терапии все пациенты данной группы находились на пресимптоматической стадии болезни. Гендерное соотношение в 1-й и 2-й группах не имело статистически значимых различий (р = 0,527).
Критерии включения:
Критерии исключения:
У всех пациентов исследовали соматический и неврологический статус, анализировали основные этапы двигательного развития с применением рекомендаций ВОЗ по 6 главным нормативным показателям достижения этапов моторного развития детей, оценивали моторные функции с применением шкал CHOP-INTEND (Тест детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорождённых), HINE-2 (шкала для короткого обследования на основе балльной системы для оценки двигательных функций детей в возрасте 2–24 мес), HFMSE (Расширенная шкала оценки моторных функций больницы Хаммерсмит), а также психо-речевое развитие, проводили электронейромиограмму, определяли уровни лёгких, средних и тяжёлых цепей нейрофиламентов в крови.
Комплексную оценку клинических, нейрофизиологических и биохимических показателей у пациентов с СМА осуществляли до инициации генной терапии и через 6, 12, 18 и 24 мес после её проведения. В данной публикации представлены результаты клинической оценки пациентов с СМА с анализом основных этапов двигательного развития.
Статистический анализ проводили с использованием программы «StatTech v. 4.8.0» («Статтех»).
Количественные показатели оценивали на предмет соответствия нормальному распределению с помощью критерия Шапиро–Уилка (при числе исследуемых менее 50).
Количественные показатели, выборочное распределение которых соответствовало нормальному, описывали с помощью средних арифметических величин (M) и стандартных отклонений (SD). В качестве меры репрезентативности для средних значений указывали границы 95% доверительного интервала (ДИ). В случае отсутствия нормального распределения количественные данные описывали с помощью медианы (Me) и нижнего и верхнего квартилей [Q1; Q3].
Категориальные данные описывали с указанием абсолютных значений (n) и долей (%), минимума (min) и максимума (max).
Сравнения в группе трёх и более параметров по количественному показателю, распределение которого отличалось от нормального, выполняли с помощью критерия Краскела–Уоллиса, попарные сравнения — с помощью критерия Данна с поправкой Холма. Различия считали статистически значимыми при p < 0,05.
На проведение данного исследования было получено разрешение локального этического комитета ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России.
Результаты
Дети с СМА I типа (1-я группа)
У абсолютного большинства пациентов 1-й группы — 43 (97,7%) было 2 копии гена SMN2, у 1 (2,3%) — 3 копии гена SMN2.
Возраст дебюта клинических симптомов в 1-й группе составил 1,15 ± 1,1 (95% ДИ 0,81–1,49), min — с рождения, max — 4,5.
Все дети с СМА I типа имели задержку формирования моторных навыков. До инициации терапии оценка по шкале HINE-2 составляла: Ме 2,0 (1,00–2,25) балла, оценка по шкале CHOP-INTEND: Ме 30,0 (22,00–37,25) баллов.
Все пациенты 1-й группы получили лечение ОА. Средний возраст введения препарата составил 3,61 ± 1,90 мес (95% ДИ 3,04–4,19), min — 1,00, max — 7,00.
Средний возраст начала лечения детей 1-й группы, диагноз которым был установлен до внедрения расширенного неонатального скрининга, составил 4,95 ± 1,33 мес (95% ДИ 4,37–5,54), min — 2,00, max — 7,00; детей, диагноз СМА которым был установлен в рамках расширенного неонатального скрининга, — 2,27 ± 1,35 мес (95% ДИ 1,67–2,87), min — 1,00, max — 6,00.
К моменту завершения набора материала в исследование (март 2025 г.) из 44 детей 1-й группы 37 детей находились под наблюдением в течение не менее 12 мес, 26 детей — в течение 18 мес, 23 ребёнка — в течение 24 мес после генной терапии ОА.
Дети с СМА I типа после проведения генной терапии демонстрировали положительную динамику в двигательном развитии (рис. 1). К концу 2-го года наблюдения согласно своему возрасту хорошо удерживали голову 39 (88,6%) детей; переворачивались из положения лёжа на спине на живот 39 (88,6%) детей; сидели без опоры 26 (60,5%) детей и только 3 (10,3%) ребёнка смогли сформировать все двигательные навыки, однако ни один из этих детей не сформировал их в соответствии с критериями ВОЗ.
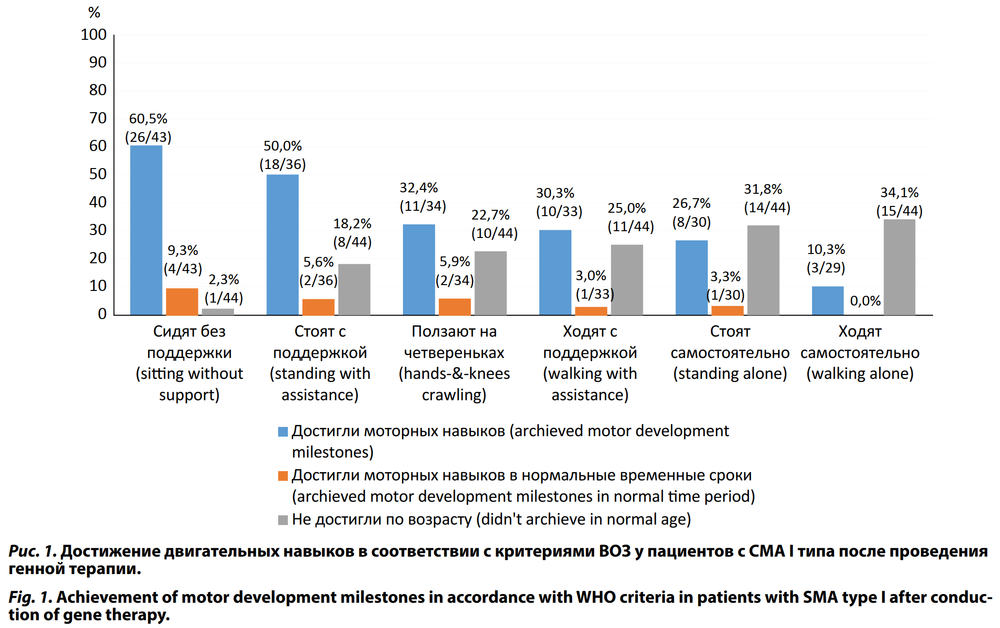
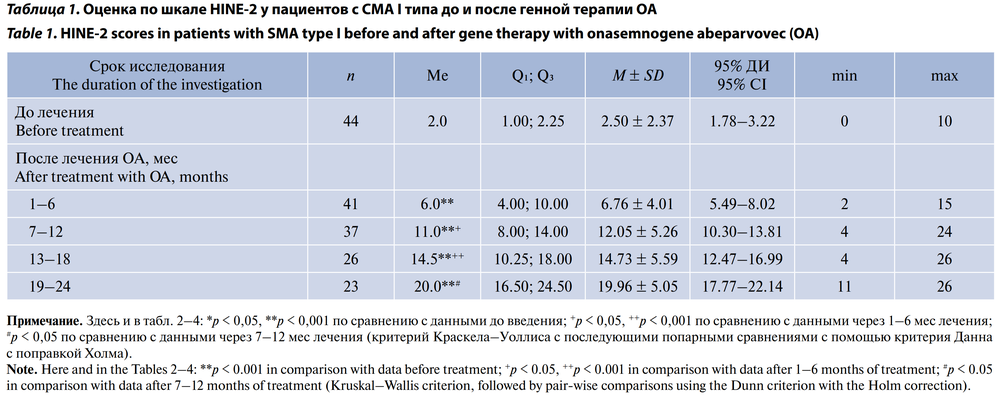
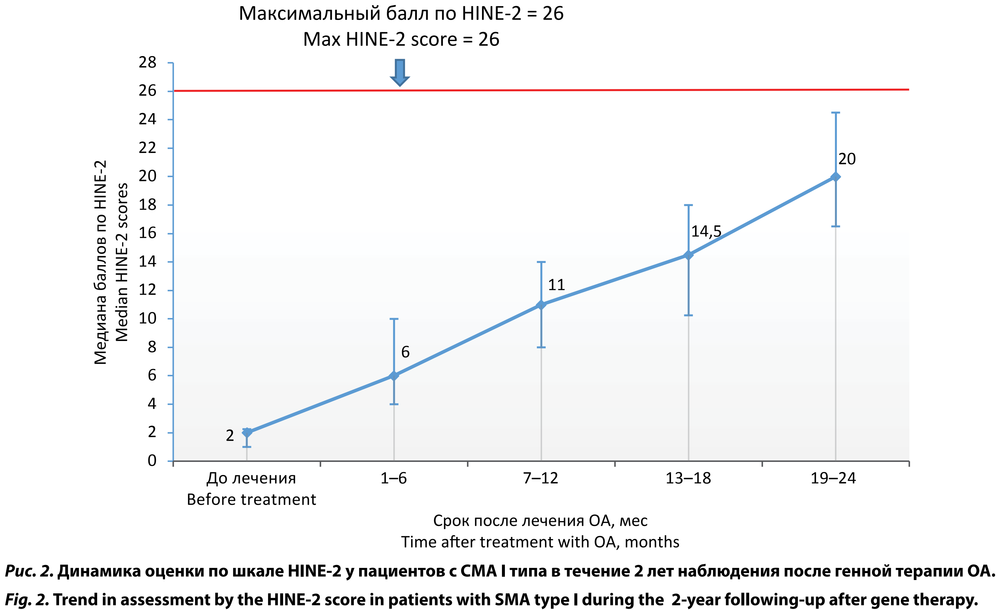
К концу периода наблюдения средняя оценка по шкале HINE-2 составила 19,96 ± 5,05 (95% ДИ 17,77–22,14) балла. Максимального значения 26 баллов достигло только 2 (4,5%) детей 1-й группы (табл. 1, рис. 2).
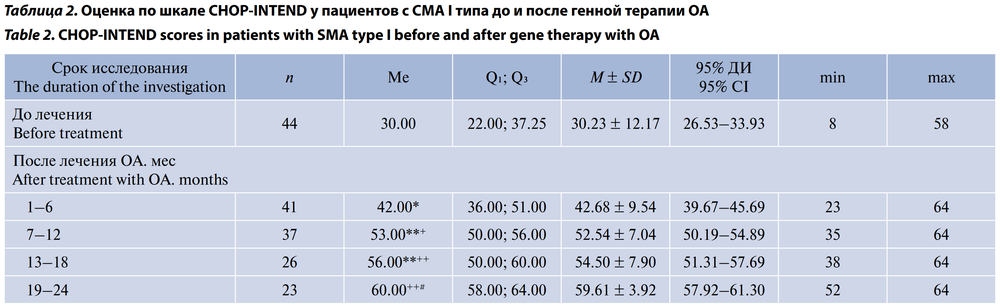
По шкале CHOP-INTEND по окончании периода наблюдения среднее значение было равно 59,61 ± 3,93 (95% ДИ 57,92–61,30) балла. Только 5 (11,4%) детей 1-й группы достигли максимального значения 64 балла к концу 2-го года наблюдения (табл. 2; рис. 3).
Дети с СМА без развития симптомов заболевания (2-я группа)
У абсолютного большинства пациентов 2-й группы — 31 (88,6%) было 3 копии гена SMN2, у 4 (11,4%) — 2 копии гена SMN2.
Возраст введения ОА во 2-й группе составил 2,0 ± 0,94 мес (95% ДИ 1,68–2,32), min — 1,00, max — 5,00.
К моменту завершения набора материала в исследование (март 2025 г.) из 35 детей 2-й группы 30 детей находились под наблюдением в течение минимум 12 мес после генной терапии препаратом ОА, 23 ребёнка — 18 мес, 12 детей — 24 мес. Такое распределение пациентов по времени наблюдения в нашем исследовании связано с тем, что неонатальный скрининг на СМА стартовал с января 2023 г. и дети на пресимптоматической стадии болезни включались по мере их выявления при молекулярно-генетической диагностике.
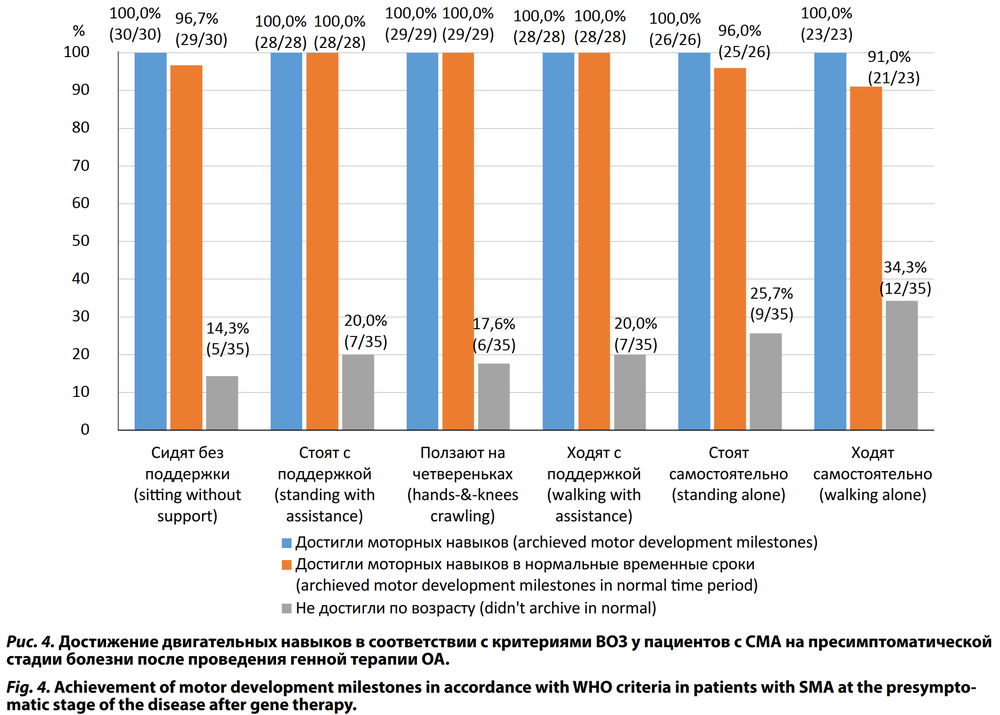
Все дети 2-й группы, получившие генную терапию ОА на пресимптоматической стадии болезни, в дальнейшем достигли почти всех двигательных навыков по критериям ВОЗ соответственно своему возрасту (рис. 4). Однако 1 (3,3%) ребёнок из 2-й группы достиг навыка «сидит самостоятельно без поддержки» и «стоит самостоятельно» позже — в 10 и 18 мес соответственно и 2 ребёнка достигли навыка «ходит самостоятельно» в 20 и 21 мес соответственно (рис. 4).
До терапии оценку двигательной активности по шкалам HINE-2 и CHOP-INTEND проводили всем детям, повторную оценку — в зависимости от сроков госпитализации детей после проведённого лечения.
При оценке по шкале HINE-2 перед стартом терапии ОА во 2-й группе среднее значение показателя составило 4,71 ± 4,15 (95% ДИ 3,29–6,14). Все дети из этой группы, которые находились под наблюдением и достигли возраста самостоятельного хождения (23 ребёнка), имели максимальное значение 26 баллов по шкале HINE-2 к возрасту 18 мес (табл. 3). Динамика по шкале HINE-2 представлена на рис. 5.
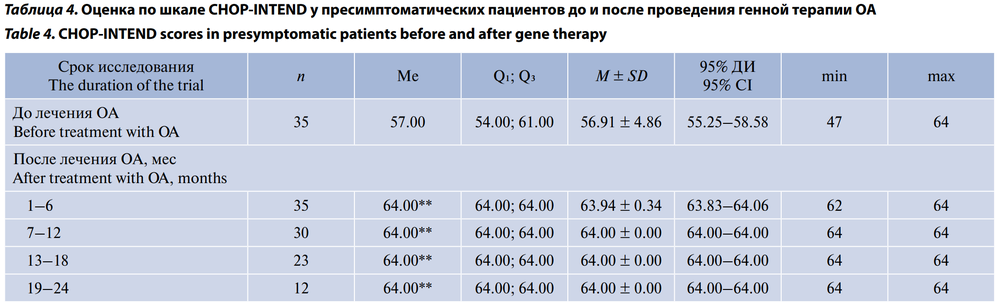
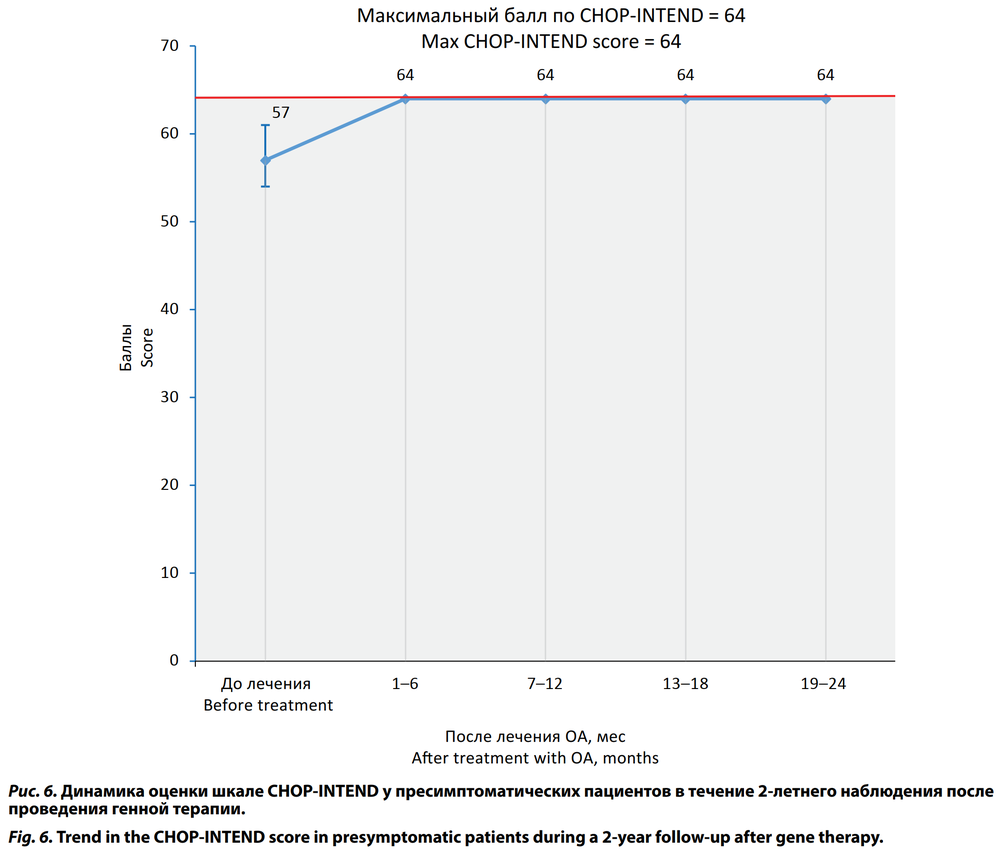
Среднее значение по шкале CHOP-INTEND перед стартом терапии ОА во 2-й группе составило 56,91 ± 4,86 (95% ДИ 55,25–58,58) балла. Через 6 мес после генной терапии ОА все дети этой группы достигли максимального значения 64 балла (табл. 4; рис. 6).
Обсуждение
Наше наблюдение представляет наиболее крупный массив данных из одного центра (44 ребёнка с СМА I типа и 35 детей на пресимптоматической стадии болезни) по расширенной оценке (основные этапы двигательного развития, шкалы HINE-2 и CHOP-INTEND при катамнезе до 24 мес наблюдения после лечения) в условиях реальной клинической практики для пациентов с СМА, которые получали монотерапию ОА.
В нашем исследовании ключевые этапы раннего моторного развития были достигнуты у большинства пациентов с СМА I типа: 88,6% детей хорошо удерживали голову и переворачивались из положения лёжа на спине на живот; 60,5% сидели без опоры. Но только 10,3% смогли сформировать все двигательные навыки, однако ни один из этих детей не сформировал их в соответствии с критериями ВОЗ.
Эти результаты оказались даже лучше, чем в европейском исследовании STR1VE-EU [13], где самостоятельное сидение в течение как минимум 10 с было достигнуто у 15 (45%) из 33 пациентов. Наши результаты показывают, что лечение ОА приводит к значительному улучшению двигательной активности при СМА I типа и самостоятельное сидение достижимо у большинства пациентов.
Оценка по шкале HINE-2 у пациентов с СМА I типа также претерпела значимые изменения и к концу периода наблюдения (через 24 мес после введение препарата ОА) составила (Ме) 20,0 (16,50–24,50) баллов. Но только 2 (4,5%) детей этой группы достигли максимальной оценки 26 баллов. В исследовании на французской популяции у пациентов с СМА I типа с 2 копиями гена SMN2 на фоне генной терапии также было показано достоверное увеличение баллов по шкале HINE-2 по сравнению с исходным тестированием (Ме) 4,0 (0–9) баллов): 7,0 (5,5–8,5) баллов через 6 мес, 11,5 (8,4–13,6) — через 12 мес, 14,8 (12,1–18,0) через 24 мес [14]. Необходимо подчеркнуть, что при естественном течении болезни дети с СМА I типа почти никогда не увеличивают оценку по шкале HINE-2, а при наблюдении через 6 мес и более отмечается её снижение у всех пациентов [15].
Показатели по шкале CHOP-INTEND до лечения у пациентов с СМА I типа в нашем исследовании были несколько выше, чем в европейском исследовании STR1VE-EU (Ме) — 28,0 (22–32) баллов [13], но ниже, чем в американском исследовании STR1VE-US (Ме) — 33,5 (24–38) баллов [16]. При этом все 44 пациента с СМА I типа, включённые в наше исследование, показали значимое улучшение двигательных функций при оценке по шкале CHOP-INTEND по сравнению с фоновым обследованием. У пациентов, участвовавших в исследованиях STR1VE-US и STR1VE-EU, также наблюдалось быстрое и устойчивое улучшение показателей по шкале CHOP-INTEND на фоне лечения ОА, хотя максимальные показатели в исследовании STR1VE-EU были несколько ниже — (Ме) 49,5 (48–52) баллов, что может быть связано с меньшими показателями на исходном этапе и несколько более коротким периодом наблюдения [13]. Несмотря на эти различия, степень улучшений по сравнению с исходными уровнями была схожей. Также следует отметить, что при естественном течении болезни дети с СМА I типа почти никогда не достигают оценку по шкале CHOP-INTEND больше 40 баллов к возрасту 6 мес [6, 17, 18].
Полученные нами данные согласуются с результатами большинства зарубежных исследований о высокой эффективности ОА у пациентов с СМА I типа, что приводит к существенной модификации течения заболевания [14, 16, 19, 20].
Несмотря на достоверные улучшения в двигательной сфере, дети с СМА I типа имели через 2 года после генной терапии ОА значительные ограничения в самостоятельном передвижении. Как показывает исследование французских авторов, применение ОА при СМА I типа не смогло предотвратить развитие деформации позвоночника у таких детей [14]. Это подчёркивает тот факт, что, хотя генная терапия значительно улучшает двигательные показатели при СМА I типа, но пациенты по-прежнему могут страдать от физически значимых осложнений и инвалидизации.
Большинство пресимптоматических пациентов в нашем исследовании на фоне генной терапии достигли всех двигательных навыков по критериям ВОЗ соответственно своему возрасту. Только 1 (3,3%) ребёнок из этой группы достиг навыков «сидит самостоятельно без поддержки» и «стоит самостоятельно» позже — в 10 и 18 мес соответственно и 2 (6,6%) ребёнка достигли навыка «ходит самостоятельно» также позже — в 20 и 21 мес соответственно.
В исследовании SPR1NT у пресимптоматических пациентов с 3 копиями гена SMN2 из 15 детей, получавших генную терапию, 14 могли стоять без поддержки и 11 могли ходить самостоятельно при оценке достижения двигательных навыков по критериям ВОЗ [21]. При дальнейшем наблюдении было показано, что дети, не достигшие двигательных навыков по возрасту, в более старшем возрасте овладевали этими навыками.
В исследовании SPR1NT все 14 пресимптоматических пациентов с 2 копиями гена SMN2 овладели навыком самостоятельного сидения и 11 (79%) из них достигли этого двигательного рубежа в пределах нормального периода развития, установленного ВОЗ [22]. Из 14 пресимптоматических пациентов 11 (79%) могли стоять без опоры и 7 (50%) из них достигли этого навыка в рамках нормального моторного развития по ВОЗ. Ходили самостоятельно 10 (71%) из 14 детей и 6 (43%) сделали это в рамках критериев ВОЗ.
Все представленные выше результаты свидетельствуют о том, что у пресимптоматических детей, верифицированных на неонатальном скрининге, как с 3, так и с 2 копиями гена SMN2 средний возраст достижения первых моторных навыков был ниже по сравнению с пациентами с СМА I типа. Также у пресимптоматических пациентов значительно больший процент детей достигал двигательного развития по критериям ВОЗ. Эти результаты неоспоримо указывают на необходимость раннего выявления и своевременного лечения пациентов с СMA с 2 и 3 копиями гена SMN2 [14, 16, 23].
Исходные показатели и среднее общее увеличение показателей по шкале HINE-2, полученные в нашем исследовании, были значительно больше у пресимптоматических пациентов по сравнению с детьми с СМА I типа. Но даже в группах пациентов с СМА I типа с 2 и 3 копиями гена SMN2 двигательная функция, оценённая по шкале HINE-2, достоверно различались при фоновом тестировании и в возрасте 24 мес на терапии ОА [14], при этом улучшение двигательных возможностей наблюдалось и в диапазоне 12–24 мес жизни для всех пациентов.
У пресимптоматических пациентов оценка по шкале CHOP-INTEND в нашем исследовании перед стартом терапии ОА составила (Ме) 57,00 (54,00–61,00). Все дети этой группы достигли максимального значения 64 балла к возрасту 6 мес. В исследовании SPR1NT у пресимптоматических пациентов с 2 копиями гена SMN2 исходная оценка по шкале CHOP-INTEND была несколько ниже — 46,1 ± 8,8 балла, но быстро повышалась после инфузии ОА [22]. Через 1 мес после лечения оценка по шкале CHOP-INTEND увеличилась на 3,9 ± 8,3 балла, через 3 мес — на 11,2 ± 8,8 балла, через 6 мес оценка по шкале CHOP-INTEND составила (Ме) 60 (51–64) баллов. Все 14 детей в группе SPR1NT с 2 копиями гена SMN2 достигли показателя CHOP-INTEND как минимум 58 баллов к 18 мес [22]. Безусловно, сравнивать напрямую эти данные с результатами нашего исследования нельзя, т. к. у нас преобладали дети на пресимптоматической стадии болезни с 3 копиями гена SMN2, но общая тенденция совпадает.
Наши данные подтверждают результаты других исследований о том, что пациенты, выявленные с помощью неонатального скрининга, имеют более высокие исходные значения по шкале CHOP-INTEND и в целом демонстрируют более значительные улучшения [20–22, 24, 25].
Таким образом, результаты нашего исследования показали, что применение ОА у детей с СМА I типа существенно модифицирует течение заболевания и значимо улучшает исходы по сравнению с естественной историей развития СМА с дебютом в раннем возрасте, а у большинства пресимптоматических пациентов использование генной терапии приводило к достижению основных этапов двигательного развития в соответствии с критериями ВОЗ.
Наше исследование имеет ряд ограничений. В первую очередь это связано с наблюдением за пациентами в условиях реальной клинической практики, из-за чего часть пациентов не всегда строго придерживалась графика наблюдения. Вследствие этого оценка некоторых основных этапов двигательного развития у ряда детей могла носить относительно субъективный характер. Продолжительность наблюдения варьировала, особенно у пресимтоматических пациентов. Это обусловлено тем, что неонатальный скрининг на СМА стартовал в России с января 2023 г. и дети на пресимптоматической стадии болезни включались по мере их выявления при молекулярно-генетической диагностике. Таким образом, к моменту завершения набора материала в исследование (март 2025 г.) из 35 детей 2-й группы 30 детей находились под наблюдением в течение минимум 12 мес, 23 детей — в течение 18 мес, 12 детей — в течение 24 мес после проведения генной терапии ОА.
Заключение
В нашем исследовании было показано, что пациенты раннего возраста с СМА после проведения генной терапии ОА демонстрировали достоверную положительную динамику в двигательном развитии. При этом дети с СМА I типа, несмотря на сохраняющуюся задержку формирования моторных навыков, смогли достичь основных этапов раннего моторного развития у большинства пациентов, что подтверждалось значительным повышением баллов по шкалам HINE-2 и CHOP-INTEND. Пациенты на пресимптоматической стадии болезни на лечении препаратом ОА достигали всех основных двигательных навыков, причём абсолютное большинство детей этой группы формировали навыки в нормативные возрастные сроки по критериями ВОЗ, что было подтверждено высокими оценками по шкалам моторного развития для детей с нервно-мышечными заболеваниями. Таким образом, результаты нашего исследования показали, что применение ОА в лечении детей с СМА I типа и у пресимптоматических пациентов существенно модифицирует течение заболевания и значимо улучшает исходы по сравнению с естественной историей развития СМА у детей с дебютом в раннем возрасте. Эти данные указывают на необходимость максимально раннего выявления и своевременного лечения пациентов с СMA с 2 и 3 копиями гена SMN2.
1. Arnold W.D., Kassar D., Kissel J.T. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015; 51(2): 157–67. https://doi.org/10.1002/mus.24497
2. Mercuri E., Sumner C.J., Muntoni F., Darras B.T., Finkel R.S. Spinal muscular atrophy. Nat. Rev. Dis. Primers. 2022; 8(1): 52. https://doi.org/10.1038/s41572-022-00380-8
3. Calucho M., Bernal S., Alías L., March F., Venceslá A., Rodríguez-Álvarez F.J., et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018; 28(3): 208–15. https://doi.org/10.1016/j.nmd.2018.01.003
4. Mercuri E., Finkel R.S., Muntoni F., Wirth B., Montes J., Main M., et al. Diagnosis and management of spinal muscular atrophy: Part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018; 28(2): 103–15. https://doi.org/10.1016/j.nmd.2017.11.005
5. Finkel R.S., Mercuri E., Meyer O.H., Simonds A.K., Schroth M.K., Graham R.J., et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018; 28(3): 197–207. https://doi.org/10.1016/j.nmd.2017.11.004
6. Kolb S.J., Coffey C.S., Yankey J.W., Krosschell K., Arnold W.D., Rutkove S.B., et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017; 82(6): 883–91. https://doi.org/10.1002/ana.25101
7. Hoy S.M. Nusinersen: a review in 5q spinal muscular atrophy. CNS Drugs. 2021; 35(12): 1317–28. https://doi.org/10.1007/s40263-021-00878-x
8. Paik J. Risdiplam: a review in spinal muscular atrophy. CNS Drugs. 2022; 36(4): 401–10. https://doi.org/10.1007/s40263-022-00910-8
9. Yeo C.J.J., Tizzano E.F., Darras B.T. Challenges and opportunities in spinal muscular atrophy therapeutics. Lancet Neurol. 2024; 23(2): 205–18. https://doi.org/10.1016/s1474-4422(23)00419-2
10. Al-Zaidy S.A., Kolb S.J., Lowes L., Alfano L.N., Shell R., Church K.R., et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J. Neuromuscul. Dis. 2019; 6(3): 307–17. https://doi.org/10.3233/jnd-190403
11. Naveed A., Calderon H. Onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J. Pediatr. Pharmacol. Ther. 2021; 26(5): 437–44. https://doi.org/10.5863/1551-6776-26.5.437
12. Петрухин А.С. Развитие нервной системы у новорожденных и детей раннего возраста. Методика исследования. Синдромы поражения. В кн.: Петрухин А.С. Детская неврология. Том 1. М.: ГЭОТАР-Медиа; 2018: 199–221.
13. Mercuri E., Muntoni F., Baranello G., Masson R., Boespflug-Tanguy O., Bruno C., et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021; 20(10): 832–41. https://doi.org/10.1016/s1474-4422(21)00251-9
14. Desguerre I., Barrois R., Audic F., Barnerias C., Chabrol B., Davion J.B., et al. Real-world multidisciplinary outcomes of onasemnogene abeparvovec monotherapy in patients with spinal muscular atrophy type 1: experience of the French cohort in the first three years of treatment. Orphanet. J. Rare Dis. 2024; 19(1): 344. https://doi.org/10.1186/s13023-024-03326-3
15. Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017; 377(18): 1723–32. https://doi.org/10.1056/nejmoa1702752
16. Day J.W., Finkel R.S., Chiriboga C.A., Connolly A.M., Crawford T.O., Darras B.T., et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021; 20(4): 284–93. https://doi.org/10.1016/s1474-4422(21)00001-6
17. Finkel R.S., McDermott M.P., Kaufmann P., Darras B.T., Chung W.K., Sproule D.M., Kang P.B., Foley A.R., Yang M.L., Martens W.B., Oskoui M., Glanzman A.M., Flickinger J., Montes J., Dunaway S., O’Hagen J., Quigley J., Riley S., Benton M., Ryan P.A., Montgomery M., Marra J., Gooch C., De Vivo D.C. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014; 83(9): 810–817. https://doi.org/10.1212/WNL.0000000000000741.
18. Mercuri E., Lucibello S., Perulli M., Coratti G., de Sanctis R., Pera M.C., et al. Longitudinal natural history of type I spinal muscular atrophy: a critical review. Orphanet J. Rare Dis. 2020; 15(1): 84. https://doi.org/10.1186/s13023-020-01356-1
19. Blair H.A. Onasemnogene abeparvovec: a review in spinal muscular atrophy. CNS Drugs. 2022; 36(9): 995–1005. https://doi.org/10.1007/s40263-022-00941-1
20. Servais L., Day J.W., De Vivo D.C., Kirschner J., Mercuri E., Muntoni F., et al. Real-world outcomes in patients with spinal muscular atrophy treated with onasemnogene abeparvovec monotherapy: findings from the RESTORE registry. J. Neuromuscul. Dis. 2024; 11(2): 425–42. https://doi.org/10.3233/jnd-230122
21. Strauss K.A., Farrar M.A., Muntoni F., Saito K., Mendell J.R., Servais L., et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat. Med. 2022; 28(7): 1390–7. https://doi.org/10.1038/s41591-022-01867-3
22. Strauss K.A., Farrar M.A., Muntoni F., Saito K., Mendell J.R., Servais L., et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat. Med. 2022; 28(7): 1381–9. https://doi.org/10.1038/s41591-022-01866-4
23. Weiß C., Becker L.L., Friese J., Blaschek A., Hahn A., Illsinger S., et al. Efficacy and safety of gene therapy with onasemnogene abeparvovec in children with spinal muscular atrophy in the D-A-CH-region: a population-based observational study. Lancet Reg. Health Eur. 2024; 47: 101092. https://doi.org/10.1016/j.lanepe.2024.101092
24. Pane M., Berti B., Capasso A., Coratti G., Varone A., D’Amico A., et al. Onasemnogene abeparvovec in spinal muscular atrophy: predictors of efficacy and safety in naïve patients with spinal muscular atrophy and following switch from other therapies. EClinicalMedicine. 2023; 59: 101997. https://doi.org/10.1016/j.eclinm.2023.101997
25. Kariyawasam D.S., D’Silva A.M., Sampaio H., Briggs N., Herbert K., Wiley V., et al. Newborn screening for spinal muscular atrophy in Australia: a non-randomised cohort study. Lancet Child Adolesc. Health. 2023; 7(3): 159–70. https://doi.org/10.1016/s2352-4642(22)00342-x
Аспирант, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
у-mail: fisenko.daria@mail.ru
Доктор мед. наук, зав. лаб. нервных болезней, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Доктор мед. наук, профессор, начальник центра детской психоневрологии, зав. отделением психоневрологии и нейрореабилитации, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия; Клинический институт детского здоровья имени Н.Ф. Филатова ФГАОУ ВО «Первый МГМУ имени И.М. Сеченова» Минздрава России (Сеченовский университет), 119435, Москва, Россия
Канд. мед. наук, зав. отделением диагностики и восстановительного лечения, начальник методического аккредитационно-симуляционного центра, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва
Канд. мед. наук, врач-невролог, ст. науч. сотр. лаб. нервных болезней, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, мл. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, врач-невролог, ст. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, мл. науч. сотр. лаб. детских редких наследственных болезней ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, врач-невролог, ст. науч. сотр. ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Канд. мед. наук, врач-невролог, ФГАУ «НМИЦ здоровья детей» Минздрава России, 119991, Москва, Россия
Фисенко Д.А., Куренков А.Л., Кузенкова Л.М., Черников В.В., Увакина Е.В., Попович С.Г., Бурсагова Б.И., Абдуллаева Л.М., Курова Ю.А., Адалимова Н.С., Николенко Д.С., Глоба О.В., Андреенко Н.В. Эффективность генной терапии препаратом онасемноген абепарвовек у пациентов со спинальной мышечной атрофией раннего возраста. Неврологический журнал имени Л.О. Бадаляна. 2025;6(1):13-25. https://doi.org/10.46563/2686-8997-2025-6-1-13-25. EDN: yyenrs
Fisenko D.A., Kurenkov A.L., Kuzenkova L.M., Chernikov V.V., Uvakina E.V., Popovich S.G., Bursagova B.I., Abdullaeva L.M., Kurova J.A., Adalimova N.S., Nikolenko D.S., Globa O.V., Andreenko N.V. The efficacy of gene therapy with onasemnogene abeparvovec in spinal muscular atrophy in young patients. L.O. Badalyan Neurological Journal. 2025;6(1):13-25. (In Russ.) https://doi.org/10.46563/2686-8997-2025-6-1-13-25. EDN: yyenrs

119991, Россия, г. Москва, Ломоносовский проспект, д. 2, стр. 1
Обработка персональных данных






















